Bridging the Annotation Gap: A Comprehensive Guide to Validating Computational Enzyme Predictions for Biomedical Research
The exponential growth of genomic data has far outpaced experimental characterization, leaving public databases rife with erroneous enzyme annotations that misdirect research and drug discovery.
Bridging the Annotation Gap: A Comprehensive Guide to Validating Computational Enzyme Predictions for Biomedical Research
Abstract
The exponential growth of genomic data has far outpaced experimental characterization, leaving public databases rife with erroneous enzyme annotations that misdirect research and drug discovery. This article provides a critical roadmap for researchers and drug development professionals to navigate this challenge. It explores the severe scope of the misannotation crisis, evaluates cutting-edge computational tools from machine learning to generative models, outlines robust experimental frameworks for validation, and establishes benchmarks for comparing methodological performance. By synthesizing foundational knowledge with practical application and troubleshooting guidance, this resource aims to empower scientists to critically assess computational predictions and implement rigorous, integrated validation pipelines that enhance the reliability of enzymatic data in biomedical research.
The Misannotation Crisis: Unveiling the Scale of Erroneous Enzyme Annotations
Automated functional annotation based on sequence similarity is a fundamental practice in genomics, yet its inherent pitfalls pose a significant challenge to research and drug development. This guide provides an objective comparison of the primary annotation methods—automated sequence alignment and experimental validation—by synthesizing current data and experimental protocols. The analysis reveals that even high-confidence similarity thresholds can result in startlingly high error rates, with one study inferring a 78% misannotation rate within a specific enzyme class [1]. This article details the quantitative evidence of these errors, compares the performance of computational and experimental approaches, and provides a toolkit of validation protocols and reagents essential for researchers aiming to ensure the reliability of their functional annotations.
The exponential growth of genomic data has created a massive annotation gap. Public protein databases contain hundreds of millions of entries, but the proportion that has been experimentally characterized is vanishingly small—only about 0.3% of entries in the UniProt/TrEMBL database are manually annotated and reviewed [1]. To bridge this gap, researchers and databases heavily rely on automated annotation methods that transfer putative functions from characterized sequences to new ones based on statistical similarity [2]. While this approach enables the processing of data at scale, it carries a fundamental risk: the widespread propagation of errors when annotations are transferred without sufficient evidence or validation. This problem is particularly acute in the field of enzymology, where incorrect Enzyme Commission (EC) numbers can misdirect entire research projects, lead to flawed metabolic models, and ultimately hamper drug discovery efforts. This article objectively compares the current annotation landscape, providing researchers with the data and methodologies needed to critically assess functional predictions.
Quantitative Evidence of Widespread Misannotation
The scale of the misannotation problem is not merely theoretical; it is being rigorously documented through computational and experimental studies. The data presented below demonstrate that error rates are substantial, even when sequences share significant similarity.
Table 1: Documented Misannotation Rates in Enzymes
| Study Focus | Reported Misannotation Rate | Key Findings | Source |
|---|---|---|---|
| S-2-hydroxyacid oxidases (EC 1.1.3.15) | 78% (inferred from experiment) | Of 122 representative sequences tested, at least 78% were misannotated. Only 22.5% contained the expected protein domain. | [1] |
| General Enzyme Function Conservation | >70% | Less than 30% of enzyme pairs with >50% sequence identity had entirely identical EC numbers. Errors occurred even with BLAST E-values below 10⁻⁵⁰. | [3] |
| BRENDA Database Analysis | ~18% | Nearly 18% of all sequences in enzyme classes shared no similarity or domain architecture with experimentally characterized representatives. | [1] |
The experimental investigation into EC 1.1.3.15 revealed that the majority of misannotated sequences contained non-canonical protein domains entirely different from those in known, characterized S-2-hydroxyacid oxidases [1]. This indicates that simple similarity searches can connect sequences based on short, insignificant regions, leading to fundamentally incorrect functional assignments. Furthermore, the problem is self-perpetuating; as new sequences are annotated based on these erroneous entries, the misannotation spreads throughout databases [1].
Comparative Analysis: Automated Annotation vs. Experimental Validation
To understand the root of the problem, it is crucial to compare the methodologies and performance of automated annotation tools against the gold standard of experimental validation.
Table 2: Performance Comparison of Annotation Methods
| Aspect | Automated Annotation (Sequence-Similarity Based) | Experimental Validation (High-Throughput Screening) |
|---|---|---|
| Throughput | Very High | Medium to High |
| Speed | Rapid (minutes to hours) | Slow (days to weeks) |
| Primary Methodology | Transfer of annotation via BLAST, PSI-BLAST, or tools like PANNZER2 based on sequence alignment [2] [4]. | Recombinant expression, purification, and in vitro activity assays [5] [1]. |
| Key Advantage | Enables annotation of massive datasets at low cost. | Provides direct, empirical evidence of function. |
| Key Limitation | Prone to error propagation; cannot confirm actual catalytic activity. | Lower throughput, requires specialized equipment and expertise. |
| Reliability for Critical Applications | Low to Moderate. Requires expert oversight and confirmation [6]. | High. Considered the benchmark for accuracy. |
| Handling of VUS (Variants of Uncertain Significance) | Performs poorly; tools show significant limitations with VUS interpretation [6]. | Essential for definitive classification. |
A core finding from recent evaluations of automated variant interpretation tools is that while they demonstrate high accuracy for clearly pathogenic or benign variants, they show significant limitations with variants of uncertain significance (VUS) [6]. This underscores that automation, while useful for clear-cut cases, struggles with the nuanced interpretations that often constitute the frontier of research. Expert oversight remains indispensable when using these tools in a clinical or research context [6].
Experimental Protocols for Validating Enzyme Function
To address annotation uncertainty, researchers must employ experimental validation. The following workflow and detailed protocol describe a robust method for high-throughput functional screening of putative enzymes.
Detailed Experimental Protocol
This protocol is adapted from high-throughput studies that successfully identified misannotation in enzyme classes [5] [1].
Step 1: Sequence Selection & Domain Architecture Analysis Select a diverse set of representative sequences from the enzyme class of interest, ensuring coverage of different taxonomic groups and similarity levels. In parallel, computationally analyze the Pfam domain architecture of each sequence. This pre-screen can immediately flag sequences lacking the critical catalytic domains found in experimentally characterized enzymes [1].
Step 2: Gene Cloning & Recombinant Expression Clone the genes into a suitable expression vector (e.g., pET series) and transform into an expression host like E. coli. A key consideration is sequence truncation: carefully define the boundaries of the mature protein to avoid removing essential regions (e.g., dimer interfaces) or including signal peptides that can interfere with heterologous expression, a noted pitfall in early screening rounds [5]. Induce protein expression and harvest the cells.
Step 3: Protein Purification Purify the recombinant proteins using affinity chromatography (e.g., His-tag purification). Assess the purity and concentration via SDS-PAGE and spectrophotometry. The goal is to obtain a purified, soluble protein fraction for testing.
Step 4: In Vitro Functional Assay Develop a specific, sensitive assay for the predicted catalytic activity. For oxidases like EC 1.1.3.15, this can be a spectrophotometric assay that measures the production of a colored product (e.g., a 2-oxoacid) over time [1]. Include appropriate controls:
- Negative Control: Assay buffer without the enzyme.
- Positive Control: A well-characterized enzyme with the same predicted activity. Test the purified proteins against the predicted substrate and, if possible, a panel of related substrates to check for potential alternative activities.
Step 5: Data Analysis & Validation An enzyme is considered "experimentally successful" if it can be expressed and folded in the host system and demonstrates activity significantly above background in the in vitro assay [5]. Sequences that fail this test are strong candidates for misannotation.
The Scientist's Toolkit: Research Reagent Solutions
Successful validation of enzyme function relies on a set of key reagents and computational tools. The following table details essential components for a functional annotation pipeline.
Table 3: Essential Research Reagents and Tools for Annotation & Validation
| Item Name | Function/Application | Relevance to Annotation Validation |
|---|---|---|
| pET Expression Vectors | High-level protein expression in E. coli. | Standardized system for recombinant production of uncharacterized sequences for functional testing. |
| Affinity Chromatography Resins (e.g., Ni-NTA) | Purification of recombinant His-tagged proteins. | Enables rapid purification of multiple putative enzymes for high-throughput activity screening. |
| Spectrophotometer / Microplate Reader | Kinetic measurement of enzyme activity. | Essential for running quantitative in vitro assays (e.g., measuring oxidation of substrates). |
| Phobius | Prediction of signal peptides and transmembrane domains. | Pre-experiment tool to prevent expression failures by ensuring correct sequence truncation [5]. |
| Pfam Database | Database of protein families and domains. | Critical for checking if a putatively annotated sequence contains the expected functional domains [1]. |
| BRENDA Database | Comprehensive enzyme resource. | Source of known enzymatic reactions, substrates, and characterized sequences for positive controls and rule definition. |
| PANNZER2 / Blast2GO | Automated functional annotation tools. | Represents the class of tools whose predictions require experimental validation; useful for generating initial hypotheses [2] [4]. |
The reliance on automated, sequence-similarity-based annotation is an undeniable bottleneck in genomics, leading to a documented misannotation rate that can exceed 70% for some enzyme classes. As this comparison has shown, computational tools are powerful for generating hypotheses but are not a substitute for experimental validation, especially for variants of uncertain significance.
The future of accurate functional annotation lies in integrating robust computational methods with high-throughput experimental screening. Advances in AI and machine learning are beginning to incorporate multiple lines of evidence beyond simple sequence alignment, such as protein structure and genomic context, which may improve predictions [7]. Furthermore, the development of high-throughput experimental platforms makes it increasingly feasible to validate predictions on a family-wide scale [5] [1]. For researchers in drug development and basic science, a critical and evidence-based approach to functional annotations is not just best practice—it is a necessity to ensure the integrity and reproducibility of their work.
The exponential growth of genomic sequence data has vastly outpaced the capacity for experimental protein characterization, making computational annotation a cornerstone of modern biology. However, the reliability of these automated annotations remains a critical concern for researchers in basic science and drug development. As of 2024, the UniProtKB/Swiss-Prot database contains only 0.64% of manually annotated enzyme sequences amidst 43.48 million total enzyme sequences, creating substantial reliance on computational function transfer [8]. This dependency creates a propagation pipeline where initial misannotations perpetuate throughout databases, potentially compromising metabolic models, drug target identification, and engineering applications. A large-scale community-based assessment (CAFA) revealed that nearly 40% of computational enzyme annotations are erroneous [8], highlighting the systemic nature of this problem. This guide examines a definitive case study quantifying misannotation rates, explores experimental validation methodologies, and objectively compares computational tools that aim to address these critical challenges.
The EC 1.1.3.15 Case Study: Experimental Revelation of Widespread Misannotation
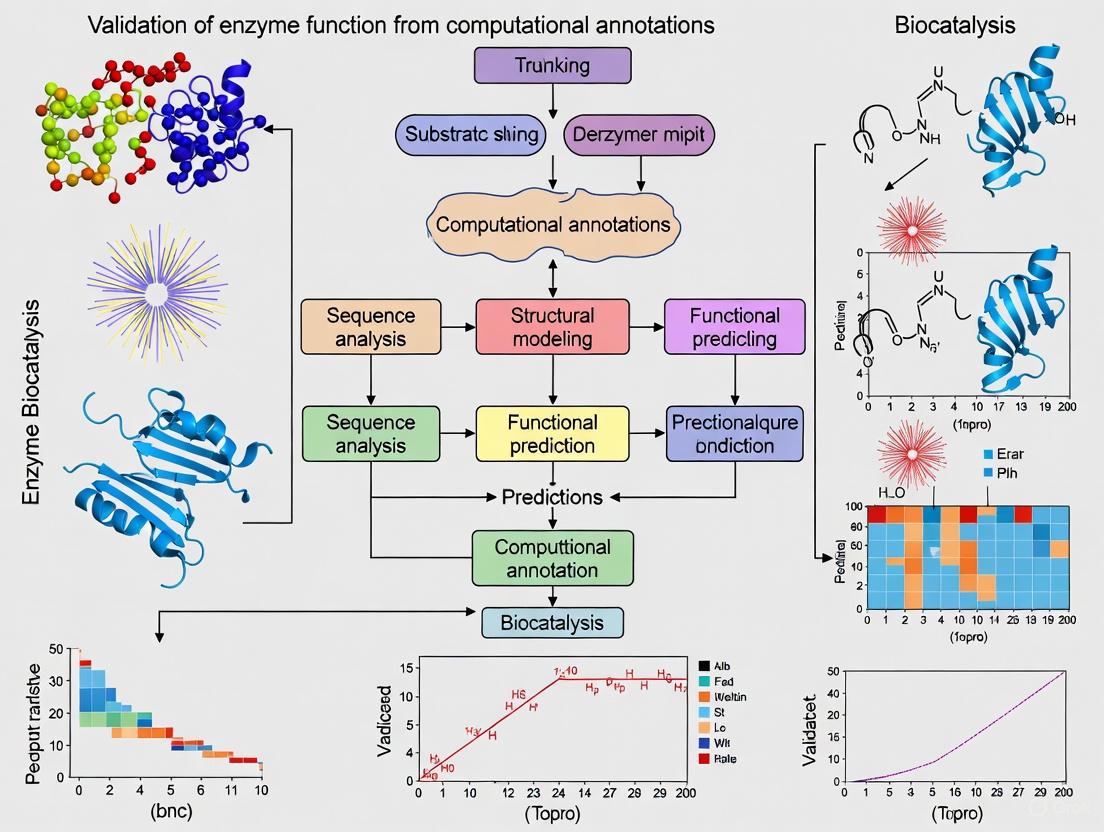
A landmark 2021 investigation conducted an experimental validation of annotations for the S-2-hydroxyacid oxidase enzyme class (EC 1.1.3.15), selected as a proof-of-concept model [9] [1]. This class catalyzes the oxidation of S-2-hydroxyacids like glycolate or lactate to 2-oxoacids using oxygen as an electron acceptor, with importance in photorespiration, fatty acid oxidation, and human health [1]. Researchers employed a high-throughput experimental platform to systematically verify predicted functions through a multi-stage validation workflow (Figure 1).
Figure 1: Experimental workflow for validating enzyme function annotations
Pre-Experimental Bioinformatics Analysis
Before experimental validation, computational analysis of the 1,058 unique sequences annotated to EC 1.1.3.15 revealed concerning patterns:
- Limited characterized diversity: Only 17 sequences had experimental evidence, with 14 of eukaryotic origin, indicating severe characterization bias [1].
- Low sequence similarity: 79% of sequences shared less than 25% sequence identity with any characterized S-2-hydroxyacid oxidase [9] [1].
- Non-canonical domain architectures: Only 22.5% contained the expected FMN-dependent dehydrogenase domain (PF01070), while others contained divergent domains including FAD-binding and 2Fe-2S binding domains [1].
Table 1: Pre-experimental Sequence Analysis of EC 1.1.3.15
| Analysis Parameter | Finding | Implication |
|---|---|---|
| Characterized Sequences | 17/1058 (1.6%) | Extreme reliance on computational annotation |
| Sequence Identity to Characterized | 79% <25% identity | Low similarity for reliable homology transfer |
| Canonical Domain Presence | 22.5% with FMN_dh domain | Majority lack essential functional domains |
| Taxonomic Distribution | >90% bacterial | Limited diversity of characterized sequences |
Experimental Findings and Misannotation Quantification
Functional screening of the 65 soluble proteins provided definitive evidence of widespread misannotation:
- Overall misannotation rate: At least 78% of sequences in the enzyme class were misannotated [9] [1].
- Alternative activities: Researchers confirmed four distinct alternative enzymatic activities among the misannotated sequences [1].
- Temporal pattern: Misannotation within the enzyme class increased over time, demonstrating error propagation [1].
- Broader implications: Computational analysis extended to all enzyme classes in BRENDA revealed that nearly 18% of all sequences were annotated to enzyme classes while sharing no similarity or domain architecture with experimentally characterized representatives [9].
Table 2: Experimental Validation Results for EC 1.1.3.15
| Experimental Metric | Result | Significance |
|---|---|---|
| Selected Representatives | 122 sequences | Covered diversity of sequence space |
| Soluble Proteins | 65/122 (53%) | Archaeal/eukaryotic less soluble |
| Misannotation Rate | ≥78% | Experimental confirmation of error rate |
| Alternative Activities | 4 confirmed | Misannotations represent real functional diversity |
| BRENDA-wide Problem | ~18% of sequences | Misannotation affects even well-studied classes |
Experimental Protocols for Enzyme Function Validation
High-Throughput Functional Screening Methodology
The experimental platform employed for validating EC 1.1.3.15 annotations provides a template for systematic enzyme function verification:
Gene Selection and Synthesis: Selected 122 representative sequences covering the diversity of EC 1.1.3.15 sequence space, with consideration of taxonomic origin, domain architecture, and similarity clusters [1].
Recombinant Expression: Cloned and expressed genes in Escherichia coli using high-throughput protocols. Achieved soluble expression for 65 proteins (53%), with archaeal and eukaryotic proteins proportionally less soluble [9].
Activity Assay: Screened soluble proteins for S-2-hydroxyacid oxidase activity using the Amplex Red peroxide detection assay [9]. This fluorometric method detects hydrogen peroxide production, a byproduct of the oxidase reaction.
Alternative Activity Identification: For misannotated sequences, employed broader substrate profiling to identify correct functions, discovering four alternative activities [1].
Computational Validation Methods
Complementary bioinformatic analyses provide orthogonal validation:
- Domain Architecture Analysis: Used Pfam to identify functional domains; canonical FMN-dependent dehydrogenase domain (PF01070) expected for genuine S-2-hydroxyacid oxidases [1].
- Sequence Similarity Networks: Visualized functional relationships and identified non-isofunctional subgroups [10].
- Active Site Analysis: Compared active site residues with characterized enzymes to identify functionally critical conservation [10].
Research Reagent Solutions for Enzyme Validation
Table 3: Essential Research Reagents for Enzyme Function Validation
| Reagent/Resource | Function in Validation | Application Example |
|---|---|---|
| Amplex Red Peroxide Detection | Fluorometric detection of H₂O₂ production | Oxidase activity screening [9] |
| BRENDA Database | Comprehensive enzyme function resource | Reference for EC classifications [9] |
| Pfam Database | Protein domain family annotation | Identifying canonical functional domains [1] |
| UniProtKB/Swiss-Prot | Manually curated protein database | High-quality reference sequences [8] |
| Gene Synthesis Services | Custom DNA sequence production | Expressing diverse enzyme variants [1] |
| Solubility Screening | Assessing recombinant protein expression | Filtering functional candidates [9] |
Next-Generation Computational Tools: Performance Comparison
To address annotation challenges, new machine learning tools have emerged with different architectural approaches and performance characteristics (Table 4).
Table 4: Comparison of Enzyme Function Prediction Tools
| Tool | Approach | Input Data | Key Advantages | Performance Notes |
|---|---|---|---|---|
| SOLVE [8] | Ensemble (RF, LightGBM, DT) with focal loss | Primary sequence (6-mer tokens) | Interpretable via Shapley analyses; distinguishes enzymes/non-enzymes | Optimized for class imbalance; high accuracy across EC levels |
| CLEAN-Contact [11] | Contrastive learning + contact maps | Sequence + structure (contact maps) | Integrates sequence and structure information | 16.22% higher precision than CLEAN on New-3927 dataset |
| EZSpecificity [12] | SE(3)-equivariant GNN | 3D enzyme structures | Specificity prediction from active site geometry | 91.7% accuracy vs. 58.3% for previous model on halogenases |
| ProteInfer [11] | Convolutional neural network | Primary sequence | End-to-end prediction from sequence | Lower precision than CLEAN-Contact in benchmarks |
| DeepEC [11] | Neural networks | Primary sequence | Specialized for EC number prediction | Lower performance on rare EC numbers |
Performance Benchmarking Insights
Independent evaluations reveal significant performance variations:
- CLEAN-Contact demonstrates 2.0- to 2.5-fold improvement in precision compared to DeepEC, ECPred, and ProteInfer on the Price-1497 dataset [11].
- SOLVE effectively handles class imbalance through focal loss penalty and provides interpretability via Shapley analysis identifying functional motifs [8].
- EZSpecificity shows remarkable accuracy (91.7%) in identifying single potential reactive substrates for halogenases, significantly outperforming previous methods (58.3%) [12].
- Current ML models still mostly fail to make novel predictions and can make basic logic errors that human annotators avoid, underscoring the need for uncertainty quantification [10].
Implications for Research and Development
Impact on Biological Research and Drug Development
The high misannotation rates have profound implications:
- Metabolic Modeling: Incorrect enzyme annotations compromise genome-scale metabolic models, affecting predictions of metabolic capabilities [11].
- Drug Target Identification: Human HAO1 (a genuine EC 1.1.3.15 enzyme) is a target for primary hyperoxaluria; misannotations could divert research efforts [1].
- Enzyme Engineering: Reliable function prediction is crucial for designing microbial cell factories for medicine, biomanufacturing, and bioremediation [11].
- Microbiome Studies: Gut microbiota enzymes require accurate annotation, as alterations are associated with inflammatory bowel disease and obesity [8].
Recommendations for Researchers
To enhance annotation reliability in research workflows:
- Experimental Validation: Prioritize functional assays for high-value targets, using the described high-throughput platform as a template.
- Tool Selection: Choose computational tools based on specific needs: SOLVE for interpretable sequence-based prediction, CLEAN-Contact for maximum accuracy, EZSpecificity for substrate specificity predictions.
- Multi-Method Consensus: Employ multiple prediction tools and be skeptical of low-confidence annotations, particularly for rare EC numbers.
- Domain Architecture Verification: Always check for canonical functional domains when assigning enzyme functions.
- Consideration of Paralog Complexity: Account for non-isofunctional paralogous groups, a common source of misannotation [10].
The 78% misannotation rate in EC 1.1.3.15 serves as a powerful reminder of the fundamental challenges in enzyme bioinformatics. While next-generation computational tools show promising improvements in accuracy, the gold standard remains experimental validation. Researchers must approach computational annotations with appropriate skepticism, implement robust validation strategies, and stay informed of rapidly evolving prediction technologies to ensure biological conclusions and therapeutic applications rest on firm functional foundations.
Incorrect enzyme functional annotations represent a critical and widespread challenge in biochemical research and drug discovery. These errors, stemming largely from automated sequence-based annotation transfer, lead to significant resource waste, project delays, and increased safety risks in pharmaceutical development. Recent studies reveal that approximately 78% of sequences in some enzyme classes may be misannotated, with nearly 18% of all enzyme sequences in major databases sharing no similarity to experimentally characterized representatives [1] [9]. This comprehensive analysis examines the tangible consequences of these annotations and provides experimental frameworks for validation, offering researchers practical solutions to mitigate risks in their projects.
The Scale and Scope of the Misannotation Problem
Quantitative Evidence of Widespread Misannotation
The problem of enzyme misannotation is not isolated but systemic across biological databases. Experimental validation efforts reveal alarming statistics about annotation reliability:
Table 1: Documented Enzyme Misannotation Rates Across Studies
| Study Focus | Misannotation Rate | Sample Size | Primary Method |
|---|---|---|---|
| S-2-hydroxyacid oxidases (EC 1.1.3.15) | 78% | 122 sequences | High-throughput experimental screening [1] |
| All enzyme classes in BRENDA | 18% of sequences lack similarity to characterized representatives | Entire database analysis | Computational analysis of domain architecture & similarity [9] |
| Bacterial sequences in EC 1.1.3.15 | 79% share <25% sequence identity to characterized sequences | 1,058 sequences | Sequence identity analysis [9] |
The misannotation problem extends beyond single enzyme families. Computational analysis of the BRENDA database reveals that nearly one-fifth of all annotated sequences lack meaningful similarity to experimentally characterized representatives, suggesting systematic issues in annotation pipelines [9]. This problem has practical consequences: researchers may spend months studying proteins with completely incorrect functional assignments, derailing projects before they even begin.
Root Causes of Erroneous Annotations
Understanding the origins of misannotation is crucial for developing solutions. The primary drivers include:
- Automated annotation transfer: Most proteins have functions assigned automatically through sequence similarity to curated entries, with only 0.3% of UniProt/TrEMBL entries manually reviewed [1] [9]
- Convergent evolution limitations: Proteins with similar functions may have low sequence similarity, while those with different functions can share high sequence similarity [13]
- Non-canonical domain architectures: In the S-2-hydroxyacid oxidase class, only 22.5% of sequences contained the canonical FMN-dependent dehydrogenase domain, with the majority having divergent domain structures [9]
- Taxonomic bias: Characterized sequence diversity is limited, with 14 of 17 characterized enzymes in EC 1.1.3.15 being of eukaryotic origin, creating representation gaps [9]
Direct Consequences for Drug Discovery and Development
Impact on Pharmacological Profiling and Safety Assessment
In drug discovery, incorrect enzyme annotations directly impact safety assessment and compound optimization. Recent analysis of pharmacological profiling practices reveals systematic underrepresentation of nonkinase enzymes in safety panels [14]:
Table 2: Enzyme Representation in Pharmacological Profiling
| Profiling Aspect | Finding | Implication |
|---|---|---|
| Enzyme inclusion rate | ~25% of studies included no enzymes in selectivity profiling | Critical safety gaps for compounds targeting non-enzyme targets [14] |
| Overall enzyme representation | Only 11% of targets in pharmacological screens are enzymes | Disconnect from therapeutic reality, as enzymes comprise ~33% of FDA-approved drug targets [14] |
| Hit rate significance | Enzymes have comparable or higher hit rates in selectivity screens | Undetected off-target effects pose clinical safety risks [14] |
This underrepresentation creates significant safety gaps. When investigational molecules target non-enzyme targets, the proportion of enzymes in selectivity screens falls below average, creating blind spots for off-target effects [14]. Given that enzymes constitute the largest pharmacological target class for FDA-approved drugs (approximately one-third of all targets), this discrepancy is particularly concerning [14].
Economic and Timeline Consequences
The downstream effects of misannotation carry substantial economic impacts:
- Late-stage failures: Toxicity observed in clinical trials accounts for 30% of drug candidate failures [14]
- Resource misallocation: Projects pursuing incorrectly annotated targets waste significant R&D resources before errors are detected
- Extended timelines: The need for later target validation and correction of erroneous pathways delays project milestones
The recent FDA analysis of Investigational New Drug applications confirms that enzymes are tested less frequently than other molecular targets despite having comparable or higher hit rates in selectivity screens, indicating a systematic blind spot in current safety assessment practices [14].
Experimental Validation Frameworks
High-Throughput Experimental Validation
To address the misannotation crisis, researchers have developed robust experimental frameworks for validation. A comprehensive approach for validating S-2-hydroxyacid oxidase annotations demonstrates a scalable methodology [1] [9]:
Figure 1: Experimental Validation Workflow for enzyme function.
This experimental pipeline successfully identified that 78% of sequences annotated as S-2-hydroxyacid oxidases were misannotated, with four distinct alternative activities confirmed among the misannotated sequences [1] [9]. The methodology highlights that approximately 53% of expressed proteins were soluble, with archaeal and eukaryotic proteins showing proportionally lower solubility than bacterial counterparts [9].
Computational Validation Metrics
For computationally generated enzymes, recent research has established benchmarking frameworks to evaluate sequence functionality before experimental validation. The Computational Metrics for Protein Sequence Selection (COMPSS) framework improves the rate of experimental success by 50-150% by combining multiple evaluation metrics [5]:
Table 3: Computational Metrics for Predicting Enzyme Functionality
| Metric Category | Examples | Strengths | Limitations |
|---|---|---|---|
| Alignment-based | Sequence identity, BLOSUM62 scores | Detects general sequence properties | Ignores epistatic interactions, position equality [5] |
| Alignment-free | Protein language model likelihoods | Fast computation, no homology requirement | May miss structural constraints [5] |
| Structure-based | Rosetta scores, AlphaFold2 confidence | Captures atomic-level function determinants | Computationally expensive for large sets [5] |
Experimental validation of over 500 natural and generated sequences demonstrated that only 19% of tested sequences were active, highlighting the critical need for improved computational filters before experimental investment [5]. Key failure modes included problematic signal peptides, transmembrane domains, and disruptive truncations at protein interaction interfaces [5].
AI and Advanced Computational Solutions
Machine Learning and Deep Learning Approaches
Artificial intelligence methods are increasingly addressing the limitations of traditional sequence-similarity approaches:
- Conventional machine learning: Algorithms like k-Nearest Neighbors (kNN), Support Vector Machines (SVM), and Random Forests using features from amino acid compositions and physicochemical properties [13]
- Deep neural networks: Complex architectures that learn patterns directly from raw amino acid sequences without extensive feature engineering [13]
- Transformer-based models: Leveraging self-attention mechanisms to capture long-range dependencies in protein sequences [13]
These methods overcome critical limitations of traditional approaches, particularly in handling cases where convergent evolution creates proteins with similar functions but low sequence similarity, or where divergent evolution results in proteins with different functions but high sequence similarity [13].
Experimental Validation of AI-Generated Enzymes
Rigorous evaluation of AI-generated enzymes reveals both promise and limitations. Assessment of sequences produced by three contrasting generative models (ancestral sequence reconstruction, generative adversarial networks, and protein language models) showed varying success rates [5]:
- Ancestral sequence reconstruction: Generated 9 of 18 active copper superoxide dismutases and 10 of 18 active malate dehydrogenases
- Generative adversarial networks: Only 2 of 18 active copper superoxide dismutases and 0 of 18 active malate dehydrogenases
- Language models: 0 of 18 active sequences for both enzyme families
These results highlight that while computational generation can produce novel sequences, robust experimental validation remains essential, as model performance varies significantly by approach [5].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 4: Key Research Reagents for Enzyme Validation Studies
| Reagent/Solution | Function | Application Example |
|---|---|---|
| Recombinant expression systems (E. coli) | Heterologous protein production | High-throughput expression of 122 candidate sequences [9] |
| Amplex Red peroxide detection | Fluorometric activity detection | Screening for S-2-hydroxyacid oxidase activity [9] |
| Phobius prediction tool | Signal peptide and transmembrane domain identification | Filtering out sequences with problematic domains [5] |
| BRENDA Database | Reference enzyme functional data | Benchmarking against experimentally characterized sequences [1] [9] |
| UniProt/TrEMBL | Comprehensive sequence database | Source of diverse sequences for analysis [1] [9] |
| Pfam domain architecture analysis | Protein domain identification | Detecting non-canonical domain arrangements [9] |
| AlphaFold2 | Protein structure prediction | Residue confidence scoring for structure-based metrics [5] |
Incorrect enzyme annotations represent a critical vulnerability in biomedical research and drug discovery pipelines, with demonstrated potential to derail projects through misdirected resources, safety oversights, and late-stage failures. The documented 78% misannotation rate in some enzyme classes, coupled with the systematic underrepresentation of enzymes in pharmacological profiling, creates perfect conditions for project failure [14] [1] [9].
Moving forward, the field requires:
- Integrated validation pipelines that combine computational pre-screening with medium-throughput experimental verification
- Expanded pharmacological profiling panels that better represent enzyme targets, particularly nonkinase enzymes
- Database curation initiatives to address systematic annotation errors
- Improved AI models with demonstrated experimental validation
By adopting rigorous validation frameworks and recognizing the limitations of current annotations, researchers can mitigate these risks and build more reliable drug discovery pipelines. The consequences of incorrect annotations are too significant to ignore, affecting everything from basic research conclusions to clinical trial outcomes and patient safety.
The exponential growth of genomic data has profoundly transformed biological research, yet this abundance of sequence information presents a significant challenge: the reliability of functional annotations. With nearly 185 million entries in the UniProt/TrEMBL protein database and only 0.3% manually annotated and reviewed in Swiss-Prot, the vast majority of proteins have their function assigned through automated methods [1] [9]. This over-reliance on computational inference has led to a crisis of misannotation that permeates public databases and compromises research validity. A groundbreaking experimental investigation into a single enzyme class (EC 1.1.3.15) revealed that at least 78% of sequences were incorrectly annotated, with the majority containing non-canonical protein domains and lacking predicted activity [1] [9]. This startling finding underscores the critical limitation of sequence-based annotation and highlights the urgent need for approaches that integrate domain architecture and three-dimensional active site structure for accurate functional validation.
The pervasiveness of this problem extends across enzyme classes, with a computational analysis of the BRENDA database indicating that nearly 18% of all sequences are annotated to enzyme classes while sharing no similarity or domain architecture to experimentally characterized representatives [1] [9]. This misannotation crisis affects even well-studied enzyme classes of industrial and medical relevance, potentially leading research astray and hampering drug discovery efforts. As we move further into the structural genomics era, with initiatives like the AlphaFold Database releasing over 214 million predicted structures [15], the scientific community faces both unprecedented opportunities and formidable challenges in bridging the gap between sequence data and biological function.
Experimental Evidence: Quantifying the Misannotation Crisis
Case Study: Systematic Misannotation in EC 1.1.3.15
A rigorous experimental investigation of the S-2-hydroxyacid oxidase enzyme class (EC 1.1.3.15) provides compelling evidence for the misannotation crisis. Researchers selected 122 representative sequences spanning the diversity of this enzyme class for experimental validation [1] [9]. Through high-throughput synthesis, cloning, and recombinant expression in Escherichia coli, they obtained 65 soluble proteins (53% solubility rate) for functional characterization [9]. The experimental workflow involved testing each soluble protein for S-2-hydroxy acid oxidase activity using the Amplex Red peroxide detection system, which provides a sensitive spectrophotometric readout of enzymatic function.
The results revealed a startling discrepancy between computational predictions and experimental evidence. While all selected sequences were annotated as EC 1.1.3.15 enzymes, the majority lacked the predicted activity. Analysis of sequence similarity and domain architecture provided crucial insights into the root causes of misannotation. Notably, 79% of sequences annotated as EC 1.1.3.15 shared less than 25% sequence identity with the closest experimentally characterized representative [1] [9]. Even more telling was the domain architecture analysis, which showed that only 22.5% of the 1,058 sequences in this enzyme class contained the canonical FMN-dependent dehydrogenase domain (FMN_dh, PF01070) characteristic of genuine 2-hydroxy acid oxidases [1] [9]. The majority were predicted to contain non-canonical domains, including FAD binding domains characteristic of entirely different oxidoreductase families, cysteine-rich domains, and 2Fe-2S binding domains, suggesting fundamentally different biochemical functions.
Beyond a Single Enzyme Class: The Broader Implications
The misannotation problem identified in EC 1.1.3.15 is not an isolated case but rather representative of a systemic issue affecting functional databases. Experimental confirmation of four alternative enzymatic activities among the misannotated sequences demonstrates how erroneous annotations can obscure true biological function and hinder the discovery of novel enzymatic activities [1] [9]. Furthermore, the study documented that misannotation within this enzyme class has increased over time, suggesting that the problem is compounding as automated annotation pipelines process ever-larger datasets without sufficient experimental validation [1].
This misannotation crisis has real-world consequences for biotechnology and medicine. Enzymes in the EC 1.1.3.15 class play crucial roles in various biological processes and applications: plant glycolate oxidases are essential for photorespiration, mammalian hydroxyacid oxidases participate in glycine synthesis and fatty acid oxidation, and bacterial lactate oxidases are used in clinical biosensors for lactate monitoring in healthcare and sports medicine [1] [9]. Human HAO1 has been proposed as a therapeutic target for treating primary hyperoxaluria, a metabolic disorder causing renal decline [1] [9]. Inaccurate annotations of such medically relevant proteins could significantly impede drug discovery efforts and the development of diagnostic tools.
Table 1: Experimental Validation of EC 1.1.3.15 Annotations
| Analysis Parameter | Finding | Implication |
|---|---|---|
| Sequences tested | 122 representative sequences | Comprehensive coverage of sequence diversity |
| Solubility rate | 65 soluble proteins (53%) | Representative functional testing |
| Misannotation rate | ≥78% of sequences | Widespread incorrect functional assignments |
| Sequence similarity | 79% with <25% identity to characterized enzymes | Limited basis for homology-based transfer |
| Domain architecture | Only 22.5% contain canonical FMN-dependent dehydrogenase domain | Fundamental structural mismatch with annotation |
| Alternative activities | 4 confirmed among misannotated sequences | True functions being obscured by incorrect annotations |
Computational Methods: From Sequence to Structure
Limitations of Sequence-Based Annotation
Traditional automated annotation methods primarily rely on sequence similarity to infer function, an approach that fails to account for the complex relationship between sequence, structure, and function. The fundamental assumption that sequence similarity implies functional similarity breaks down at lower identity levels, particularly below the "twilight zone" of 25% sequence identity where structural and functional divergence becomes common [1]. This limitation is exacerbated by the exponential growth of sequence databases, which outpaces the capacity for experimental characterization and creates a propagation cycle where misannotations beget further misannotations.
Recent research has demonstrated that even advanced generative protein sequence models struggle to predict functional enzymes reliably. In a comprehensive evaluation of computational metrics for predicting in vitro enzyme activity, researchers expressed and purified over 500 natural and generated sequences with 70-90% identity to natural sequences [5]. The initial round of "naive" generation resulted in mostly inactive sequences, with only 19% of experimentally tested sequences showing activity above background levels [5]. This poor performance highlights the limitations of sequence-centric approaches and underscores the need for structural validation, particularly for distinguishing functional enzymes from non-functional counterparts with similar sequences.
Structural Alignment and Comparison Tools
The emergence of massive structural databases has driven the development of advanced structural alignment algorithms capable of efficiently comparing three-dimensional protein structures. Traditional structural alignment methods like DALI and CE provided accurate comparisons but required several seconds per structure pair, making them impractical for large-scale analyses [15]. More recent approaches have focused on improving computational efficiency while maintaining accuracy through innovative strategies.
The SARST2 algorithm represents a significant advancement in structural alignment technology, employing a filter-and-refine strategy that integrates primary, secondary, and tertiary structural features with evolutionary statistics [15]. This method utilizes machine learning with decision trees and artificial neural networks to rapidly filter out non-homologous structures before performing more computationally intensive detailed alignments. In benchmark evaluations, SARST2 achieved an impressive 96.3% accuracy in retrieving family-level homologs, outperforming other state-of-the-art methods including FAST (95.3%), TM-align (94.1%), and Foldseek (95.9%) [15]. Notably, SARST2 completed searches of the massive AlphaFold Database significantly faster than both BLAST and Foldseek while using substantially less memory, enabling researchers to perform large-scale structural comparisons on ordinary personal computers [15].
Table 2: Performance Comparison of Structural Alignment Methods
| Method | Accuracy | Speed | Memory Efficiency | Key Features |
|---|---|---|---|---|
| SARST2 | 96.3% | Fastest | Highest | Integrated filter-and-refine strategy with machine learning |
| Foldseek | 95.9% | Fast | Moderate | 3Di structural strings from deep learning |
| FAST | 95.3% | Moderate | Moderate | Pioneering rapid structural alignment |
| TM-align | 94.1% | Moderate | Moderate | Widely used for topology-based alignment |
| BLAST | 89.8% | Slow (sequence-based) | Low | Sequence-only comparison |
Integrated Workflows: Combining Computational and Experimental Approaches
The COMPSS Framework for Predicting Enzyme Functionality
The development of the Composite Metrics for Protein Sequence Selection (COMPSS) framework represents a significant step forward in predicting enzyme functionality from sequence and structural features. Through three rounds of iterative experimentation and computational refinement, researchers established a composite computational metric that improved the rate of experimental success by 50-150% compared to naive selection methods [5]. This framework evaluates sequences using a combination of alignment-based, alignment-free, and structure-supported metrics to account for various factors that influence protein folding and function.
The COMPSS framework was rigorously validated using two enzyme families—malate dehydrogenase (MDH) and copper superoxide dismutase (CuSOD)—selected for their substantial sequence diversity, physiological significance, and complex multimeric structures [5]. The evaluation included sequences generated by three contrasting generative models: ancestral sequence reconstruction (ASR), generative adversarial networks (GANs), and protein language models (ESM-MSA) [5]. This comprehensive approach demonstrated that no single metric could reliably predict function, but an appropriately weighted combination could significantly enhance the selection of functional sequences for experimental testing.
High-Throughput Experimental Validation Platforms
Advanced experimental platforms are essential for validating computational predictions at scale. The high-throughput pipeline used for characterizing EC 1.1.3.15 enzymes exemplifies this approach, incorporating gene synthesis, cloning, recombinant expression in E. coli, solubility assessment, and enzymatic activity assays [1] [9]. This systematic workflow enables rapid functional screening of hundreds of sequences, generating crucial experimental data to benchmark and refine computational predictions.
For structural characterization, integrative approaches combine multiple experimental techniques to elucidate the hierarchical organization of protein structures:
- Primary structure: Determined through classical sequencing techniques (Edman degradation, dansyl chloride assays) complemented by modern mass spectrometry-based peptide mapping and emerging nanopore-based sequencing technologies [16]
- Secondary structure: Probed using circular dichroism spectroscopy to quantify α-helical and β-sheet content, monitor conformational transitions, and evaluate thermostability [16]
- Tertiary structure: Resolved through high-resolution methods including X-ray crystallography, nuclear magnetic resonance spectroscopy, and cryo-electron microscopy, often complemented by computational predictions [16]
This multi-level structural analysis provides crucial insights into active-site geometry, protein-protein interactions, and functional divergence within protein superfamilies, enabling researchers to move beyond sequence-based annotations to understand the structural determinants of function.
Diagram 1: Integrated workflow for validating enzyme function, combining computational analysis with experimental validation.
Research Reagent Solutions: Essential Tools for Enzyme Validation
Table 3: Essential Research Reagents and Platforms for Enzyme Function Validation
| Reagent/Platform | Function | Application in Validation |
|---|---|---|
| Amplex Red Peroxide Detection | Spectrophotometric detection of peroxide production | Functional assay for oxidase activity [1] [9] |
| ESM-MSA Transformer | Protein language model for sequence generation | Generating novel sequences for functional testing [5] |
| ProteinGAN | Generative adversarial network for protein sequence design | Creating diverse sequences beyond natural variation [5] |
| Ancestral Sequence Reconstruction | Statistical phylogenetic model | Resurrecting ancient sequences with enhanced stability [5] |
| SARST2 Structural Aligner | Rapid structural alignment against massive databases | Identifying structural homologs and functional domains [15] |
| Circular Dichroism Spectroscopy | Secondary structure quantification | Assessing proper protein folding and stability [16] |
| AlphaFold2 | AI-based structure prediction | Generating 3D models for active site analysis [5] [15] |
The validation of enzyme function requires moving beyond sequence-based assumptions to incorporate domain architecture and three-dimensional active site structure. Experimental evidence demonstrates that 78% of sequences in at least one enzyme class are misannotated when relying solely on sequence similarity [1] [9], highlighting the critical importance of structural validation. The development of integrated computational and experimental workflows, such as the COMPSS framework [5] and advanced structural alignment tools like SARST2 [15], provides researchers with powerful methodologies for accurate functional annotation.
As structural databases continue to expand, with initiatives like the AlphaFold Database now containing over 214 million predicted structures [15], the research community has unprecedented resources for structural and functional analysis. By leveraging these resources alongside high-throughput experimental validation platforms, researchers can overcome the limitations of traditional annotation methods and advance our understanding of the complex relationship between protein structure and function. This integrated approach is essential for accelerating drug discovery, understanding disease mechanisms, and harnessing the full potential of genomic data for biomedical innovation.
Computational Arsenal: From AI-Driven Prediction to Generative Design
The accurate computational annotation of enzyme function represents a critical challenge at the intersection of bioinformatics and drug development. With the UniProt/TrEMBL database containing nearly 185 million entries and only 0.3% manually annotated and reviewed, the research community heavily relies on automated function prediction, which can result in significant error rates [1]. Experimental validation of the S-2-hydroxyacid oxidase enzyme class (EC 1.1.3.15) revealed that at least 78% of sequences were misannotated, highlighting the severity of this problem [1]. This validation crisis in enzyme annotation has created an urgent need for more sophisticated computational approaches that can reliably predict enzyme function before costly experimental verification.
Cross-attention graph neural networks (GNNs) have emerged as a powerful framework for addressing this challenge by simultaneously modeling multiple data modalities and relationships. These architectures extend beyond conventional GNNs by incorporating attention mechanisms that enable dynamic weighting of features from different sources—such as node features, topological information, and relational data—allowing for more nuanced and specific predictions of enzyme function and activity [17] [18].
Cross-Attention GNN Architectures for Biological Prediction
Graph Topology Attention Networks (GTAT)
The Graph Topology Attention Networks (GTAT) framework enhances graph representation learning by explicitly incorporating topological information through cross-attention mechanisms. GTAT operates through two sequential processes: first, it extracts topology features from the graph structure and encodes them into topology representations using Graphlet Degree Vectors (GDV), which capture the distribution of nodes in specific orbits of small connected subgraphs [17]. Second, it processes both node and topology representations through cross-attention GNN layers, allowing the model to dynamically adjust the influence of node features and topological information [17].
This architecture specifically addresses limitations of previous GNN approaches that failed to adequately integrate richer topological features beyond basic information like node degrees or edges. By treating node feature representations and extracted topology representations as separate modalities, GTAT achieves more robust expression of graph structures [17]. Experimental results demonstrate that this approach mitigates over-smoothing issues and increases robustness against noisy data, both critical factors in biological network inference [17].
Cross-Attention for Gene Regulatory Networks (XATGRN)
The Cross-Attention Complex Dual Graph Embedding Model (XATGRN) addresses the specific challenge of inferring gene regulatory networks with skewed degree distribution, where some genes regulate multiple others (high out-degree) while others are regulated by multiple factors (high in-degree) [18]. This architecture employs a cross-attention mechanism to focus on the most informative features within bulk gene expression profiles of regulator and target genes, enhancing the model's representational power for predicting regulatory relationships and their directionality [18].
XATGRN utilizes a fusion module based on Cross-Attention Network (CAN) that processes gene expression data for regulator gene R and target gene T to generate queries, keys, and values for the cross-attention mechanism [18]. The model retains half of each gene's original self-attention embedding and half of its cross-attention embedding, enabling it to handle intrinsic features of each gene while capturing complex regulatory interactions between them [18].
Configuration Cross-Attention for Tensor Compilers (TGraph)
While not directly applied to biological prediction, the TGraph architecture demonstrates the versatility of cross-attention GNNs for complex optimization problems. TGraph employs cross-configuration attention that enables explicit comparison between different configurations within the same batch, transforming the problem from individual prediction to learned ranking [19]. This approach has shown significant performance improvements, increasing mean Kendall's τ across layout collections from 29.8% to 67.4% compared to reliable baselines [19].
Performance Comparison: Cross-Attention GNNs vs. Alternative Approaches
Table 1: Performance comparison of graph neural network architectures across different domains
| Model Architecture | Application Domain | Key Metric | Performance | Reference |
|---|---|---|---|---|
| GTAT (Graph Topology Attention) | General Graph Representation | Classification Accuracy | Outperforms state-of-the-art methods on various benchmark datasets | [17] |
| XATGRN | Gene Regulatory Network Inference | Regulatory Relationship Prediction | Consistently outperforms state-of-the-art methods across various datasets | [18] |
| MPNN | Chemical Reaction Yield Prediction | R² Value | 0.75 (Highest among GNN architectures tested) | [20] |
| GAT/GATv2 | Chemical Reaction Yield Prediction | R² Value | Lower than MPNN | [20] |
| Traditional Heuristic Compilers | Tensor Program Optimization | Kendall's τ | 29.8% | [19] |
| TGraph (Cross-Attention) | Tensor Program Optimization | Kendall's τ | 67.4% | [19] |
Table 2: Experimental results for enzyme function prediction and validation
| Study Focus | Experimental System | Key Finding | Impact | Reference |
|---|---|---|---|---|
| Enzyme Annotation Accuracy | S-2-hydroxyacid oxidases (EC 1.1.3.15) | 78% misannotation rate in enzyme class | Highlights critical need for improved prediction methods | [1] |
| Computational Filter Development | Malate dehydrogenase (MDH) & Copper superoxide dismutase (CuSOD) | Improved experimental success rate by 50-150% | Demonstrates value of computational pre-screening | [5] |
| Generative Model Comparison | Ancestral sequence reconstruction, GAN, Protein Language Model | ASR generated 9/18 (CuSOD) and 10/18 (MDH) active enzymes | Establishes benchmark for generative protein models | [5] |
Experimental Protocols and Methodologies
Enzyme Function Validation Workflow
Experimental validation of computational predictions follows a rigorous multi-stage process. For enzyme function validation, this typically involves: (1) selecting representative sequences from the enzyme class; (2) synthesizing, cloning, and recombinantly expressing proteins in systems like Escherichia coli; (3) assessing protein solubility and stability; and (4) testing predicted activity through specific biochemical assays [1]. For S-2-hydroxyacid oxidases, the Amplex Red peroxide detection system serves as a key assay method, detecting hydrogen peroxide production as a byproduct of the oxidase reaction [1].
The COMPSS (Composite Metrics for Protein Sequence Selection) framework provides a structured approach for evaluating computational metrics for predicting enzyme activity. This involves multiple rounds of experimentation, starting with naive generation and progressively refining metrics based on experimental outcomes [5]. Critical parameters assessed include alignment-based metrics (sequence identity, BLOSUM62 scores), alignment-free methods (protein language model likelihoods), and structure-supported metrics (Rosetta-based scores, AlphaFold2 confidence scores) [5].
Cross-Attention GNN Implementation
Implementing cross-attention GNNs for biological prediction requires specific architectural considerations. The GTAT framework utilizes the Orbit Counting Algorithm (OCRA) to compute Graphlet Degree Vectors with time complexities of O(n·d³) and O(n·d⁴) for graphlets with up to four and five nodes respectively, where n is the number of nodes and d is the maximum node degree [17]. The topology representations are then normalized and processed through a multilayer perceptron before being input into the graph cross-attention layers [17].
For XATGRN, the cross-attention mechanism is implemented through projection matrices that map gene expression data for regulator and target genes into query, key, and value representations [18]. The model employs multi-head self-attention and cross-attention mechanisms, with each gene retaining half of its original self-attention embedding and half of its cross-attention embedding to balance intrinsic features and interaction patterns [18].
Diagram 1: Cross-attention mechanism for integrating node features and topology information in GTAT architecture
Performance Evaluation Metrics
Evaluation of enzyme function predictors utilizes multiple complementary metrics. For classification tasks, standard metrics include accuracy, precision, recall, and F1-score. The diagnostic odds ratio (DOR) serves as a combined indicator of sensitivity and specificity, providing a single metric for comparing predictive accuracy across different biomarkers or prediction methods [21]. Hierarchical summary receiver operating characteristic curves (HSROCs) account for threshold effects when summarizing overall diagnostic performance [21].
In ranking tasks such as configuration optimization, Kendall's τ correlation coefficient measures the ordinal association between predicted and actual rankings, with TGraph achieving 67.4% compared to 29.8% for traditional heuristic approaches [19]. For regression tasks including chemical reaction yield prediction, R² values quantify the proportion of variance explained by the model, with MPNN achieving 0.75 in comparative studies of GNN architectures [20].
Table 3: Key research reagents and computational resources for cross-attention GNN implementation
| Resource Category | Specific Tools/Reagents | Function/Purpose | Application Example |
|---|---|---|---|
| Experimental Validation Systems | Escherichia coli expression system | Recombinant protein production | Heterologous enzyme expression for activity testing [5] |
| Amplex Red Peroxide Assay | Detection of oxidase activity | Validation of S-2-hydroxyacid oxidase function [1] | |
| Computational Datasets | BRENDA Enzyme Database | Comprehensive enzyme functional data | Source of enzyme sequences and classifications [1] |
| TpuGraphs Dataset | Runtime measurements of computational graphs | Benchmarking configuration optimization models [19] | |
| GNN Implementation Frameworks | Graphlet Degree Vectors (GDV) | Topological feature extraction | Node topology representation in GTAT [17] |
| Orbit Counting Algorithm (OCRA) | Graphlet enumeration | Computation of GDV with reduced complexity [17] | |
| Cross-Attention Network (CAN) | Multi-modal feature fusion | Integration of regulator-target gene interactions [18] | |
| Performance Assessment Tools | Diagnostic Odds Ratio (DOR) | Combined sensitivity/specificity metric | Evaluation of prediction accuracy [21] |
| Hierarchical SROC (HSROC) | Threshold-independent performance analysis | Summary of predictive performance across studies [21] | |
| Kendall's τ | Rank correlation coefficient | Assessment of configuration ranking accuracy [19] |
Diagram 2: Integrated workflow for computational prediction and experimental validation of enzyme function
Cross-attention graph neural networks represent a significant advancement in computational enzyme function prediction, addressing critical limitations of previous approaches through their ability to integrate multiple data modalities and dynamically weight feature importance. The demonstrated success of architectures like GTAT and XATGRN across diverse biological prediction tasks, coupled with the rigorous experimental validation of computational predictions, points toward a future where in silico enzyme annotation achieves substantially higher accuracy rates.
As these methods continue to evolve, integrating additional data sources such as protein structures from AlphaFold2, metabolic pathway context, and chemical reaction data will further enhance their predictive power. For researchers and drug development professionals, these advancements translate to more reliable pre-screening of enzyme candidates, reduced experimental costs, and accelerated discovery pipelines. The cross-attention paradigm, with its flexibility and performance advantages, is poised to become a cornerstone of computational enzyme function prediction in the coming years.
The engineering of novel enzymes represents a frontier in synthetic biology, with applications ranging from sustainable chemistry and biomanufacturing to therapeutic drug design [22]. While generative artificial intelligence (AI) and protein language models (pLMs) have demonstrated remarkable capability in sampling novel protein sequences, a significant challenge remains: predicting whether these computationally generated sequences will fold into stable structures and exhibit the desired catalytic function [5]. The assumption that novel sequences drawn from a distribution similar to natural proteins will be functional does not always hold true, with experimental studies revealing that initial "naive" generation can result in a majority (over 80%) of inactive sequences [5]. This guide provides a comparative analysis of current computational models and evaluation frameworks, focusing on their performance in generating and identifying functional enzyme sequences, to serve as a benchmark for researchers navigating this complex landscape.
Comparative Analysis of Generative Models and Evaluation Metrics
The performance of AI-generated enzymes is highly dependent on the choice of generative model and the computational metrics used for evaluation. Below, we compare prominent approaches based on experimental validation studies.
Table 1: Comparison of Generative Models for Enzyme Design
| Generative Model | Model Type | Reported Experimental Success Rate | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Ancestral Sequence Reconstruction (ASR) [5] | Phylogeny-based Statistical Model | ~50-55% (MDH & CuSOD) [5] | High stability; successful resurrection of ancient functions [5] | Constrained by evolutionary history; limited novel sequence exploration [5] |
| Generative Adversarial Network (ProteinGAN) [5] | Deep Neural Network (GAN) | ~0-11% (MDH & CuSOD) [5] | Potential to explore novel sequence spaces [5] | High rate of non-functional sequences; requires robust filtering [5] |
| Protein Language Model (ESM-MSA/ESM-2) [5] [23] | Transformer-based Language Model | ~55-60% (when combined with epistasis model) [23] | Learns evolutionary patterns from massive datasets; powerful for variant prediction [24] [23] | Can produce homogeneous outputs without careful fine-tuning [24] |
| Fully Computational Workflow (Rosetta) [25] | Physical & Knowledge-Based Design | High (for designed Kemp eliminases) [25] | Can create novel active sites and achieve natural-level efficiency without screening [25] | Applied primarily to well-defined model reactions |
Evaluation Metrics: From Simple Filters to Composite Scores
Selecting the right computational metrics is critical for predicting enzyme function before costly experimental work. A landmark study systematically evaluated 20 diverse metrics, leading to the development of the COMposite Metrics for Protein Sequence Selection (COMPSS) framework [5]. This filter integrates alignment-based, alignment-free, and structure-based metrics, and was shown to improve the rate of experimental success by 50% to 150% [5].
Table 2: Key Computational Metrics for Evaluating Generated Enzyme Sequences
| Metric Category | Example Metrics | Principle | Performance in Predicting Activity |
|---|---|---|---|
| Alignment-Based [5] | Sequence Identity, BLOSUM62 Score | Relies on homology to natural sequences | Good for general properties but misses epistatic interactions; moderate predictive power alone [5] |
| Alignment-Free [5] | Protein Language Model Likelihoods (e.g., from ESM) | Fast, model-internal scoring; captures co-evolutionary signals | Sensitive to pathogenic mutations; high predictive potential when combined [5] [26] |
| Structure-Based [5] | AlphaFold2 Confidence (pLDDT), Rosetta Energy Scores | Uses predicted or designed atomic structures | Captures functional constraints but can be computationally expensive; high value in composite scores [5] |
| Specialized Prediction Models [8] | SOLVE, CLEAN, DeepEC | Machine learning models trained to predict EC number or fitness from sequence | SOLVE showed high accuracy in enzyme vs. non-enzyme classification and EC number prediction [8] |
Diagram 1: The COMPSS multi-filter workflow for selecting functional enzymes.
Experimental Protocols for Validating AI-Generated Enzymes
Rigorous experimental validation is the ultimate benchmark for any computationally generated enzyme. The following protocols are standardized in the field.
Protein Expression, Purification, and In Vitro Assay
A standard workflow for validating generated sequences involves cloning the genes into expression vectors (typically in E. coli), expressing and purifying the proteins, and testing their activity in vitro [5]. A protein is considered "experimentally successful" if it can be expressed and folded solubly and demonstrates activity significantly above background in a relevant biochemical assay [5]. Key considerations include:
- Avoiding Overtruncation: For enzymes like CuSOD, truncations that remove residues critical for multimerization (e.g., at dimer interfaces) can destroy activity [5].
- Signal Peptide Handling: For proteins from certain kingdoms (e.g., bacterial CuSOD), predicting and truncating signal peptides is essential for correct expression in heterologous systems like E. coli [5].
High-Throughput Characterization in Autonomous Platforms
Cutting-edge research now integrates AI with fully automated biofoundries. One platform, leveraging the Illinois Biological Foundry (iBioFAB), automates the entire Design-Build-Test-Learn (DBTL) cycle [23]. This includes:
- Design: Using a pLM (ESM-2) and an epistasis model (EVmutation) to design initial variant libraries.
- Build: Employing a high-fidelity assembly mutagenesis method to construct variant libraries with ~95% accuracy without intermediate sequencing [23].
- Test: Fully automated modules for transformation, colony picking, protein expression, and enzyme assays.
- Learn: Using the collected data to train machine learning models for the next design cycle. This approach has successfully engineered enzymes with 16- to 26-fold improvements in activity within four weeks [23].
Diagram 2: Autonomous enzyme engineering DBTL cycle.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Reagents and Tools for AI-Driven Enzyme Engineering
| Item Name | Function/Application | Relevant Study/Model |
|---|---|---|
| ESM-2 (650M/35M parameters) [24] [23] | Core protein language model for sequence understanding, fitness prediction, and variant generation. | BIT-LLM PROTEUS [24], Autonomous Engineering Platform [23] |
| EVmutation [23] | Epistasis model identifying co-evolutionary constraints in protein families for library design. | Autonomous Engineering Platform [23] |
| AlphaFold2/3 [22] | High-accuracy protein structure prediction; used for structural evaluation of designed enzymes. | Structure-based metrics [5], Enzyme discovery [22] |
| UniProtKB / ProteinGym [24] [26] | Curated protein sequence databases and benchmarks for model training and evaluation. | SOLVE [8], ESM training [26], BIT-LLM [24] |
| COMPSS Framework [5] | A composite computational filter integrating multiple metrics to select functional sequences. | Experimental benchmarking of generative models [5] |
| Rosetta Software Suite [25] | A comprehensive toolkit for de novo enzyme design and energy-based scoring. | High-efficiency Kemp eliminase design [25] |
The field of AI-powered enzyme generation is rapidly evolving from a trial-and-error process to a disciplined engineering science. The experimental data clearly shows that while no single model is universally superior, integrated approaches that combine the strengths of generative pLMs (like ESM-2), evolutionary principles (like ASR), and robust multi-metric evaluation frameworks (like COMPSS) significantly increase the probability of experimental success [5] [23]. Future progress hinges on several key trends: a shift from single-modal to multimodal models that integrate sequence, structure, and dynamic information; the development of intelligent agents capable of autonomously running DBTL cycles; and moving beyond static structure prediction toward the dynamic simulation of enzyme function [27]. For researchers, the critical takeaway is that successful enzyme design now depends on a synergistic pipeline—combining powerful generative models with rigorous, multi-faceted computational screening and, where possible, leveraging automation to accelerate experimental validation.
In the field of functional proteomics, a significant challenge persists: directly measuring enzyme activity remains difficult and often indirect, creating a critical gap in our understanding of cellular signaling networks. While high-throughput proteomics can readily quantify protein abundance, enzyme activity cannot be simply inferred from these levels alone, as it is dynamically regulated through mechanisms such as post-translational modifications (PTMs). This limitation is particularly problematic because dysregulated enzyme activity lies at the heart of numerous complex diseases, including cancer, diabetes, and neurodegenerative disorders. The inability to efficiently map this activity on a proteome-wide scale has hindered both basic biological discovery and the development of targeted therapies.
Traditional methods for measuring enzyme activity are typically low-throughput and cannot capture the system-wide dynamics of signaling networks. This creates a pressing need for computational tools capable of bridging this gap by inferring activity from the downstream molecular footprints enzymes leave on their substrates. PTM data, especially from phosphoproteomics experiments, contains a rich source of information about the upstream enzymatic activities that created these modification patterns. Recently, innovative computational tools have emerged to decipher these patterns, offering researchers the ability to reconstruct signaling network activity from standard proteomics data. This guide provides a comprehensive comparison of these tools, focusing on their methodologies, performance, and practical application for validating enzyme function.
Computational Tools for Activity Inference: A Comparative Analysis
Several computational approaches have been developed to infer enzyme activity from PTM data, particularly phosphoproteomics. These tools vary in their underlying algorithms, the types of enzymes they can analyze, and their analytical capabilities. The following table summarizes the key features of major tools in this domain.
Table 1: Comparison of Computational Tools for Inferring Enzyme Activity from PTM Data
| Tool Name | Supported Enzymes | Core Methodology | Input Data | Unique Features | Limitations |
|---|---|---|---|---|---|
| JUMPsem [28] | Kinases, E3 Ubiquitin Ligases, HATs | Structural Equation Model (SEM) | Quantitative PTM (e.g., phospho-, ubiquitin-, acetyl-) data | Integrates public enzyme-substrate data; motif search to expand networks; handles multiple PTM types. [28] | Does not fully account for complex cross-talk between different enzymes in signaling networks. [28] |
| KEA3 [29] | Kinases | Kinase Enrichment Analysis | List of proteins or phosphorylated proteins | Upstream kinase prediction from protein lists; uses curated kinase-substrate interactions from multiple sources. [29] | Limited to kinase activity; inference based on enrichment rather than direct quantitative modeling. |
| IKAP [28] | Kinases | Not Specified | Phosphoproteomics data | Established tool for kinase activity estimation. [28] | Outperformed by JUMPsem in precision benchmarks; specific methodology not detailed. [28] |
| KSEA [28] | Kinases | Kinase Substrate Enrichment Analysis | Phosphoproteomics data | Established method for inferring kinase activity from phosphoproteomics data. [28] | Does not appear to incorporate network context or motif discovery like JUMPsem. |
| PhosphoRS (via IsobarPTM) [30] | (PTM Localization) | Localization probability scoring | MS/MS spectra | Validates PTM site localization, which is critical for accurate activity inference. [30] | Not a direct activity inference tool; focuses on prerequisite step of confident PTM site mapping. |
In-Depth Tool Profile: JUMPsem
JUMPsem is a relatively new and innovative tool designed to overcome the limitations of existing methods. It is implemented as a modular and scalable R package and is also accessible via a user-friendly R/Shiny application, making it available to both computational biologists and wet-lab scientists [28]. Its analytical process is logically structured into three key phases:
- Data Integration and Curation: The tool begins by constructing an enzyme-substrate relationship network. It pulls known relationships from public databases and can significantly expand this network (by an average of 14.7% for kinases) using an integrated motif search strategy to predict novel substrate sites [28].
- Activity Inference via Structural Modeling: The core of its functionality uses a Structural Equation Model (SEM). This algorithm integrates the curated enzyme-substrate relationships with the quantitative PTM data from mass spectrometry to compute an inferred activity score for each enzyme [28].
- Output and Visualization: The final output includes tables of inferred enzyme activities and enzyme-substrate affinity. These results can be easily visualized and explored within the accompanying Shiny app, facilitating biological interpretation [28].
Performance and Benchmarking
In direct comparative analyses, JUMPsem has demonstrated superior performance. When researchers compared it against established tools like IKAP and KSEA using human acute myeloid leukemia (AML) cell line phosphoproteomics data, JUMPsem not only recapitulated the kinase activity patterns identified by the other tools but also discovered two unique kinase activity clusters that the others missed [28]. Furthermore, a quantitative performance assessment using benchmark datasets revealed that JUMPsem achieved slightly higher precision than IKAP across various thresholds [28].
Its utility extends beyond phosphorylation. Applied to ubiquitomics and acetylomics datasets, JUMPsem successfully identified E3 ubiquitin ligases and histone acetyltransferases (HATs) with significantly altered activity under different stress conditions and across breast cancer tumor samples, respectively [28]. This demonstrates its versatility as a general-purpose tool for enzyme activity inference.
Experimental Workflows and Methodologies
Implementing these computational tools effectively requires a foundation in standardized experimental and bioinformatics protocols. The following workflow diagram and detailed methodology outline the process from sample preparation to biological insight.
Diagram 1: Experimental workflow for inferring enzyme activity from PTM data.
Detailed Experimental Protocol
The successful application of tools like JUMPsem relies on a multi-stage process, each with critical steps:
Sample Preparation and LC-MS/MS Analysis:
- Protein Extraction and Digestion: Cells or tissues are lysed under denaturing conditions. Proteins are extracted, reduced, alkylated, and digested into peptides using a protease like trypsin.
- PTM Enrichment: Given the typically low stoichiometry of PTMs, an enrichment step is crucial. For phosphoproteomics, this involves using titanium dioxide (TiO₂) or immobilized metal affinity chromatography (IMAC) to selectively bind phosphorylated peptides. For acetylated or ubiquitinated peptides, specific immunoprecipitation antibodies are used [31].
- Isobaric Labeling (Optional): For multiplexed experiments, peptides can be labeled with isobaric tags (e.g., TMT or iTRAQ) allowing for the simultaneous quantification of multiple samples in a single MS run [30].
- Liquid Chromatography and Tandem Mass Spectrometry (LC-MS/MS): The enriched peptides are separated by liquid chromatography and analyzed by a high-resolution mass spectrometer. Data-Dependent Acquisition (DDA) or Data-Independent Acquisition (DIA) methods are used to fragment peptides and generate MS/MS spectra.
Computational Data Processing (Pre-processing for JUMPsem):
- Raw Data Conversion and Peak Picking: Raw instrument files are converted to open formats (e.g., mzML) using tools like ProteoWizard. Software may be used to perform peak detection and deisotoping [30].
- Database Search and Protein Identification: Processed spectra are searched against a protein sequence database using search engines like MSFragger (often used with the Philosopher/FragPipe platform) or MaxQuant [32]. Search parameters must include the specific PTM (e.g., phosphorylation of S/T/Y) as a variable modification.
- False Discovery Rate (FDR) Control: PSM-, peptide-, and protein-level FDRs are controlled (e.g., to 1%) using target-decoy strategies, which are implemented in modern pipelines like the Philosopher toolkit [32].
- PTM Localization Validation: A critical step is to ensure the PTM site is correctly assigned. Tools like PhosphoRS (for phosphorylation) calculate a localization probability. Only sites with high confidence (e.g., probability > 0.9) should be used for quantitative analysis [30].
- Quantification: For label-free data, peptide intensities are extracted. For isobaric tag data, the reporter ion intensities in the MS/MS spectra are quantified and corrected for isotopic impurities. Software like MSstats (an R package) is often used for subsequent statistical analysis of quantitative data [29].
Activity Inference with JUMPsem:
- Input Data Preparation: The primary input for JUMPsem is a table of quantitative modified peptide data from the previous steps [28].
- Configuration: Users can run JUMPsem via its R functions or Shiny app, specifying parameters such as the enzyme-substrate databases to use and the motif search options.
- Execution and Output Analysis: JUMPsem executes its SEM algorithm to output inferred enzyme activities. Researchers then analyze these results, often by linking activated or suppressed enzymes to known biological pathways or clinical phenotypes.
A successful research project in this field relies on a combination of computational tools, databases, and experimental reagents. The following table catalogues the key resources.
Table 2: Essential Research Reagents and Resources for PTM-based Enzyme Activity Studies
| Category | Resource Name | Function and Application |
|---|---|---|
| Computational Tools | JUMPsem (R Package) | Core tool for inferring kinase, E3 ligase, and HAT activity from quantitative PTM data. [28] |
| Philosopher / FragPipe | A comprehensive, dependency-free software toolkit for processing shotgun proteomics data, from raw spectra to peptide/protein identification and quantification. [32] | |
| MSstats (R Package) | Statistical package for relative quantification of proteins and PTMs in mass spectrometry-based proteomics; ideal for downstream analysis after Philosopher. [29] | |
| PTM Databases | PhosphoSitePlus | Manually curated resource for phosphorylation, ubiquitination, and acetylation sites, providing high-confidence data for validation and hypothesis generation. [31] |
| dbPTM | Integrated database containing information on multiple PTM types from public resources and literature, useful for cross-referencing identified sites. [31] | |
| UniProtKB/Swiss-Prot | Expertly curated protein database that includes a wealth of annotated PTM information, serving as a fundamental reference. [31] | |
| Experimental Reagents | Titanium Dioxide (TiO₂) | Microsphere resin for the highly selective enrichment of phosphorylated peptides from complex peptide mixtures prior to LC-MS/MS. |
| Tandem Mass Tag (TMT) | Isobaric chemical labels allowing multiplexing (e.g., 10-16 samples) in a single MS run, improving throughput and quantitative precision. [30] | |
| Anti-pan-specific PTM Antibodies | Antibodies for enriching peptides with specific modifications (e.g., phosphorylation, acetylation, ubiquitination) for targeted proteomics studies. |
Biological Interpretation and Pathway Mapping
The ultimate goal of inferring enzyme activity is to generate testable biological hypotheses about signaling pathways and network regulation. The activity scores generated by JUMPsem and similar tools are most meaningful when mapped onto known signaling networks. The following diagram illustrates a simplified, generic kinase signaling cascade that could be reconstructed from such data.
Diagram 2: Example kinase signaling pathway reconstructed from inferred activity.
Case Study: Application in Cancer Research
The power of this approach is exemplified by its application in disease contexts. For instance, when researchers used JUMPsem to analyze phosphoproteomics data from mouse high-grade gliomas (HGG) and normal controls, the tool revealed numerous cancer-associated changes in kinase activity that were not apparent from measuring protein abundance alone [28]. This led to the identification of previously unrecognized kinase-substrate relationships and signaling pathways driving tumorigenesis.
Similarly, in the analysis of human AML cell lines, JUMPsem identified distinct clusters of differentially active kinases that were significantly enriched in disease-relevant pathways [28]. These insights, gleaned directly from the PTM data, provide a dynamic view of the signaling landscape that is more proximal to the functional state of the cell than transcript or protein abundance, offering new potential avenues for therapeutic intervention.
The advent of computational tools like JUMPsem represents a significant leap forward in functional proteomics, moving beyond static cataloging of proteins and modifications towards a dynamic understanding of enzyme activity and signaling network regulation. While tools such as KEA3, IKAP, and KSEA have laid the groundwork, JUMPsem's use of structural equation modeling, its ability to integrate and expand enzyme-substrate networks, and its applicability beyond phosphorylation to enzymes like E3 ligases and HATs, make it a powerful and versatile platform for the community [28].
Despite these advances, challenges remain. As noted in its publication, JUMPsem does not yet fully model the complex cross-talk and feedback loops inherent in cellular signaling networks, which can sometimes lead to biased activity estimates [28]. The field is rapidly evolving, with future improvements likely to incorporate more sophisticated network biology models and machine learning approaches, including artificial neural networks (ANNs) and support vector machines (SVMs), which are already being applied to other aspects of PTM prediction [31]. As these tools mature and integrate more diverse omics data, they will become indispensable for validating computational annotations of enzyme function, ultimately accelerating the pace of discovery in basic research and drug development.
The field of protein engineering is undergoing a transformative shift with the emergence of generative models capable of designing novel protein sequences. These computational approaches promise to accelerate the discovery of enzymes and therapeutic proteins by navigating the vast sequence space beyond natural variants. This guide objectively compares three contrasting methodologies—Ancestral Sequence Reconstruction (ASR), ProteinGAN, and ESM-MSA—focusing on their performance in generating functional enzymes, supported by recent experimental validations. The evaluation is framed within the broader thesis of validating enzyme function from computational annotations, providing researchers and drug development professionals with critical insights for method selection.
Ancestral Sequence Reconstruction (ASR)
ASR is a phylogeny-based statistical method that infers the most likely sequences of ancient protein ancestors from modern descendants within an evolutionary tree [33] [34]. It operates on the principle that resurrected ancestral proteins often exhibit enhanced stability and functionality, making them valuable starting points for engineering. The protocol typically involves: (1) selecting extant homologous sequences, (2) building a multiple sequence alignment (MSA), (3) computing a phylogenetic tree, and (4) reconstructing ancestral sequences using maximum likelihood or Bayesian algorithms [33]. Benchmarking against experimental phylogenies has shown that Bayesian methods incorporating rate variation infer ancestral sequences with higher phenotypic accuracy compared to maximum parsimony approaches [34].
ProteinGAN
ProteinGAN is a generative adversarial network (GAN) based on a convolutional neural network with attention mechanisms [5] [35]. The framework consists of a generator that creates novel protein sequences and a discriminator that distinguishes between natural and generated sequences. Through adversarial training, the generator learns to produce sequences that approximate the distribution of natural protein families. Recent architectural innovations, such as the Dense-AutoGAN model, incorporate dense networks and attention mechanisms to improve sequence similarity and generate variations within a smaller range from original sequences [35].
ESM-MSA
ESM-MSA is a transformer-based protein language model that leverages multiple sequence alignments [5]. While not originally designed as a generative model, it can generate new sequences through iterative masking and sampling techniques [5] [36]. The model is trained on millions of diverse protein sequences, enabling it to learn complex evolutionary constraints and structural patterns. Protein language models like ESM-MSA create embeddings—numerical representations of sequences—that capture rich biological information, which can be used for both prediction and generation tasks [36] [37].
Performance Comparison: Experimental Data
Recent large-scale experimental studies have expressed and purified hundreds of generated sequences to benchmark the functionality of enzymes produced by these models. The table below summarizes the key performance metrics for two enzyme families: malate dehydrogenase (MDH) and copper superoxide dismutase (CuSOD).
Table 1: Experimental Success Rates for Generated Enzymes
| Generative Model | MDH Activity Rate | CuSOD Activity Rate | Key Characteristics |
|---|---|---|---|
| Ancestral Sequence Reconstruction (ASR) | 10/18 (55.6%) | 9/18 (50.0%) | High experimental success; enhanced stability; phylogenetically constrained |
| ProteinGAN | 0/18 (0%) | 2/18 (11.1%) | Low initial success; requires robust filtering |
| ESM-MSA | 0/18 (0%) | 0/18 (0%) | Low initial success; requires robust filtering |
| Natural Test Sequences | 6/18 (33.3%) | 0/18 (0%)* | Baseline for comparison |
Note: The initial failure of natural CuSOD test sequences was attributed to over-truncation during cloning that removed critical dimerization domains [5].
The data reveal striking differences in model performance. ASR consistently generated a high proportion of active enzymes for both families, with success rates of 50-55.6% [5]. This aligns with the widely reported stabilizing effect of ancestral reconstruction and its utility in protein engineering [5] [34]. In contrast, ProteinGAN and ESM-MSA showed significantly lower success rates in initial rounds of testing, with most generated sequences failing to demonstrate enzymatic activity above background levels [5].
However, it is crucial to note that these outcomes were achieved without applying computational filters to pre-select the most promising generated sequences. Subsequent optimization using Composite Metrics for Protein Sequence Selection (COMPSS)—a framework incorporating alignment-based, alignment-free, and structure-based metrics—improved experimental success rates by 50-150% for sequences generated by neural networks [5].
Detailed Experimental Protocols
Enzyme Selection and Sequence Generation
The comparative study focused on two enzyme families: malate dehydrogenase (MDH) and copper superoxide dismutase (CuSOD) [5]. These were selected due to their substantial sequence diversity, available structural data, and feasibility of functional assays. Training sets comprised 6,003 CuSOD and 4,765 MDH sequences from UniProt, filtered to ensure typical domain architectures [5]. Each model generated >30,000 sequences, from which 144 were selected for experimental validation (18 per model per enzyme family, plus natural controls), maintaining 70-80% identity to the closest natural training sequence [5].
Protein Expression and Purification
Generated sequences were synthesized as DNA constructs, cloned into expression vectors, and transformed into E. coli [5]. Proteins were expressed and purified using standardized protocols. A protein was considered experimentally successful only if it could be expressed and folded in E. coli and demonstrated activity above background in in vitro enzymatic assays [5].
Functional Assessment
Enzyme activity was measured using spectrophotometric assays specific to each enzyme's function [5]. For MDH, this typically involved monitoring NADH oxidation; for CuSOD, superoxide radical dismutation was measured. These quantitative assays provided objective measures of functional success beyond mere expression and solubility.
Figure 1: Experimental Workflow for Validating Generated Enzymes
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Research Reagents for Protein Generation Studies
| Reagent / Resource | Function / Application | Example Use in Validation |
|---|---|---|
| UniProt Database | Source of natural protein sequences for model training and benchmarking | Provided 6,003 CuSOD and 4,765 MDH training sequences [5] |
| COMPSS Metrics | Computational filter combining multiple metrics to select functional sequences | Improved experimental success rates by 50-150% [5] |
| E. coli Expression Systems | Heterologous protein expression host | Standardized expression and folding assessment [5] |
| Spectrophotometric Assays | Quantitative measurement of enzyme activity | Determined functionality of expressed enzymes (e.g., MDH, CuSOD activity) [5] |
| Phylogenetic Software | Inference of evolutionary relationships for ASR | Implemented in PAML, FastML, PhyloBayes for ancestral reconstruction [34] |
| Deep Mutational Scanning | Large-scale experimental variant effect mapping | Contributed to ProteinGym benchmark datasets [38] |
Discussion and Future Directions
The experimental data reveals a fundamental trade-off between innovation and reliability in current protein generative models. ASR provides high reliability but is constrained by evolutionary history, while deep learning models like ProteinGAN and ESM-MSA offer greater exploration capacity but require sophisticated filtering to achieve functional success [5].
For researchers prioritizing a high probability of obtaining functional enzymes, particularly for challenging targets like multidomain enzymes, ASR currently offers the most reliable path. However, for projects requiring exploration of entirely novel sequence spaces beyond natural phylogeny, generative neural networks show immense potential when coupled with robust computational scoring metrics like COMPSS [5].
Future developments will likely focus on hybrid approaches that combine the strengths of these methodologies. Domain-adaptive pretraining, where general protein language models are fine-tuned on specific functional families, has already shown promise in improving feature representation for DNA-binding proteins [37]. Similarly, incorporating structural constraints from AlphaFold2 or Rosetta into generative models may enhance the probability of generating foldable, functional sequences [5] [36].
The field is progressing toward comprehensive benchmarking platforms like ProteinGym, which provides large-scale standardized assessment for fitness prediction and design [38]. As these resources mature, they will enable more systematic comparisons and accelerate the development of next-generation protein design tools that reliably bridge the gap between computational annotation and experimental function.
From In Silico to In Vitro: Designing Robust Experimental Validation Pipelines
Quantitative High-Throughput Screening (qHTS) has become a cornerstone of modern drug discovery and functional enzymology, enabling researchers to rapidly profile thousands of compounds against biological targets. Within the specific context of validating enzyme function from computational annotations, robust assay development is not merely convenient but essential. Research has revealed alarming rates of misannotation in public databases; one systematic investigation of the S-2-hydroxyacid oxidase class (EC 1.1.3.15) found that at least 78% of sequences were incorrectly annotated, with four distinct alternative activities confirmed among the misannotated sequences [1] [39]. This widespread inaccuracy means that assays developed based solely on database annotations may be measuring non-existent functions, highlighting the critical need for rigorously validated qHTS approaches that can reliably distinguish true enzymatic activity.
The transition from traditional HTS to qHTS represents a fundamental shift from qualitative screening to precise quantitative profiling. While HTS aims to identify "hits" or "actives," qHTS generates full concentration-response curves for every compound, providing rich data on potency and efficacy [40]. This quantitative nature makes it uniquely suited for challenging computational predictions, as it can reveal subtle functional characteristics and mechanism-of-action details that are invisible in binary assays. However, the value of any qHTS campaign is fundamentally dependent on the robustness of the underlying assay. A poorly developed assay will generate misleading data that perpetuates, rather than resolves, annotation errors. This guide examines best practices in assay development, compares alternative methodologies, and provides a framework for implementing qHTS that delivers reliable, reproducible data for validating enzyme function.
Foundational Principles of Biochemical Assay Development
The Assay Development Workflow
Biochemical assay development follows a structured sequence of steps that balances scientific precision with practical screening constraints. The process begins with clearly defining the biological objective—identifying the specific enzyme or target, understanding its reaction type (kinase, methyltransferase, protease, etc.), and determining what functional outcome must be measured (product formation, substrate consumption, or binding event) [41]. This initial scoping is particularly crucial when investigating enzymes with disputed or computationally-predicted functions, as the assay design must be capable of detecting the specific catalytic activity purported in the annotation.
The subsequent stages include selecting an appropriate detection method compatible with the enzyme's catalytic products, optimizing assay components (substrate concentration, buffer composition, enzyme levels, and cofactors), and rigorously validating assay performance using statistical metrics [41]. The final stages focus on scaling and automating the validated assay for high-throughput implementation, followed by data interpretation that informs structure-activity relationships and mechanism-of-action studies [41]. Throughout this process, the goal is to create a foundation for reproducibility and scalability—essential qualities for generating reliable data that can confirm or refute computational functional predictions.
Key Assay Formats and Their Applications
Biochemical assays for qHTS generally fall into two broad categories: binding assays and enzymatic activity assays. Binding assays quantify molecular interactions such as protein-ligand binding and are typically used to measure affinity (Kd), dissociation rates (koff), or competitive displacement. Common techniques include Fluorescence Polarization (FP), Surface Plasmon Resonance (SPR), and FRET-based assays [41]. While valuable for confirming interactions, binding assays do not directly measure catalytic function and may be insufficient alone for validating enzyme annotations.
Enzymatic activity assays form the core of functional validation and can be further divided into coupled/indirect assays and direct detection assays. Coupled assays utilize a secondary enzyme system to convert the product of interest into a detectable signal, providing potential signal amplification but introducing additional variables that can complicate interpretation [41]. Direct detection assays, particularly homogeneous "mix-and-read" formats, simplify workflows by directly detecting enzymatic products without separation steps or coupling reactions. Universal activity assays like the Transcreener platform, which detects common products such as ADP (for kinases) or SAH (for methyltransferases), offer broad applicability across enzyme classes and are particularly valuable for screening multiple targets within an enzyme family [41].
Table 1: Comparison of Major Biochemical Assay Formats
| Assay Format | Detection Principle | Advantages | Limitations | Best Applications |
|---|---|---|---|---|
| Fluorescence Polarization (FP) | Changes in rotational diffusion upon binding | Homogeneous format, no separation steps | Limited dynamic range, interference from fluorescent compounds | Binding assays, competitive displacement |
| TR-FRET/FRET | Energy transfer between fluorophores in proximity | High sensitivity, reduced background | Requires specific fluorophore pairing | Binding assays, protein-protein interactions |
| Coupled Enzymatic Assays | Secondary enzyme generates detectable signal | Signal amplification, established reagents | Multiple potential interference points | When direct detection is not feasible |
| Direct Detection Assays | Direct measurement of enzymatic products | Fewer steps, reduced variability, universal applicability | May require specialized reagents | Functional screening across enzyme classes |
Methodological Deep Dive: Experimental Protocols for qHTS
Protocol: Development and Validation of a Robust Biochemical Assay
Objective: To establish a validated, miniaturized biochemical assay suitable for quantitative high-throughput screening against a target enzyme, with specific application to verifying computationally-predicted enzymatic functions.
Materials and Reagents:
- Purified enzyme (preferably >90% pure, concentration to be determined during optimization)
- Natural or synthetic substrate (with storage conditions optimized for stability)
- Appropriate reaction buffer components (including salts, cofactors, and stabilizing agents)
- Detection reagents (e.g., Transcreener ADP2 assay reagents for kinases [41])
- Reference controls (known inhibitors/activators if available)
- Low-volume microplates (384-well or 1536-well format)
- Plate reader capable of chosen detection method (FI, FP, TR-FRET)
Procedure:
Initial Assay Configuration:
- Prepare a master reaction mix containing buffer, cofactors, and substrate at a concentration approximate to the expected Km value.
- Dilute the enzyme in appropriate storage buffer to create a 2X concentrated solution.
- Dispense equal volumes of reaction mix and enzyme solution to initiate reactions.
- Incubate under optimal temperature and time conditions determined from pilot experiments.
- Stop reactions (if necessary) and add detection reagents according to manufacturer protocols.
- Measure signal using pre-validated instrument settings.
Assay Optimization Phase:
- Enzyme Titration: Test a range of enzyme concentrations (typically 0.1-10 nM for enzymes with high turnover) to identify the concentration that produces a robust signal while maintaining linearity with time.
- Time Course: Measure product formation at multiple timepoints (e.g., 15, 30, 60, 90, 120 minutes) to establish the linear range of the reaction.
- Substrate Kinetics: Determine the Km apparent for the substrate by testing a range of concentrations (typically 0.1x to 10x estimated Km).
- Buffer Optimization: Systematically vary pH, ionic strength, and critical cofactors to identify optimal reaction conditions.
Validation and Quality Control:
- Calculate the Z'-factor using positive and negative controls included on each plate: Z' = 1 - [3×(σp + σn) / |μp - μn|], where σp and σn are the standard deviations of positive and negative controls, and μp and μn are their means [41].
- Establish acceptability criteria (typically Z' > 0.5 indicates excellent assay quality suitable for HTS).
- Determine signal-to-background ratio (S/B = μp / μn) and coefficient of variation (CV = σ/μ).
- Test reference compounds (if available) to confirm expected pharmacological response.
Validation in Enzyme Annotation Studies: When developing assays to verify computational annotations, include positive control enzymes with experimentally verified functions that are phylogenetically related to the target enzyme. This provides a critical benchmark for determining whether measured activity represents true physiological function or non-specific catalytic potential [1].
Protocol: Statistical Analysis and Hit Identification in qHTS
Objective: To implement robust statistical methodology for analyzing qHTS data, classifying compound activity, and controlling false discovery rates, with particular attention to verifying ambiguous enzyme annotations.
Materials and Software:
- Normalized response data from qHTS campaign
- Statistical software with nonlinear regression capabilities (R, Python, or proprietary solutions)
- Implementation of Preliminary Test Estimation (PTE) methodology [40]
Procedure:
Data Normalization:
- Normalize raw response data relative to positive control (-100%) and vehicle control (0%) using the formula: [ y = \frac{y0 - N}{I - N} \times (-100) ] where (y) is percent activity, (y0) is the raw data value, (N) is the median of the vehicle control, and (I) is the median of a positive control [40].
Dose-Response Curve Fitting:
- Fit the four-parameter Hill model to the dose-response data for each compound: [ f(x,\theta) = \theta0 + \frac{\theta1 \theta3^{\theta2}}{x^{\theta2} + \theta3^{\theta2}} ] where (x) denotes dose, (\theta0) is the lower asymptote, (\theta1) is the efficacy, (\theta2) is the slope parameter, and (\theta_3) is the ED50 [40].
- Employ robust fitting procedures (M-estimation) to minimize the influence of outliers that are common in qHTS data.
Compound Classification:
- Implement the PTE-based classification methodology that is robust to variance structure and outliers [40].
- Test H0: θ1 = 0 (no efficacy) versus Ha: θ1 ≠ 0 using appropriate significance thresholds with multiple testing corrections.
- Classify compounds based on estimated parameters and statistical significance:
- Active: H0 rejected with θ̂2 > 0, θ̂3 < xmax and |yxmax| > 10
- Inactive: H0 not rejected or θ̂2 < 0
- Marginal: Compounds neither classified as active nor inactive
False Discovery Rate Control:
- Apply PTE methodology to maintain controlled false discovery rates while maintaining good power, overcoming limitations of both conservative (low power) and liberal (high FDR) existing methods [40].
Application to Enzyme Validation: When screening enzymes with disputed annotations, apply strict statistical criteria and consider orthogonal verification for any putative "hits" that would confirm the annotation. The high misannotation rate in public databases (78% in EC 1.1.3.15) necessitates extraordinary evidence for claimed functions [1].
Comparative Analysis of qHTS Methodologies
Statistical Methodologies for qHTS Data Analysis
The statistical analysis of qHTS data presents unique challenges due to the volume of compounds screened, the presence of heteroscedasticity (variance changing with dose), and the frequent occurrence of outliers. Traditional methodologies have included the NCGC method, which fits the Hill model using ordinary least squares and classifies compounds based on parameter estimates without accounting for uncertainty, and the Parham method, which uses likelihood ratio tests but ignores underlying variance structure [40]. More recently, Preliminary Test Estimation (PTE) methodology has been developed to address these limitations, providing robustness to variance structure and outliers through M-estimation procedures.
Table 2: Comparison of qHTS Data Analysis Methodologies
| Methodology | Statistical Approach | Variance Handling | Outlier Robustness | Classification Basis | Performance |
|---|---|---|---|---|---|
| NCGC Method | Ordinary Least Squares | Assumes homoscedasticity | Not robust | Point estimates of θ parameters without uncertainty | Conservative, low power |
| Parham Method | Likelihood Ratio Test | Ignores variance structure | Not robust | Statistical significance of θ1 | Liberal, high FDR |
| PTE-Based Method | Preliminary Test Estimation with M-estimation | Robust to heteroscedasticity | Robust via M-estimation | Statistical inference with variance modeling | Balanced FDR control and power |
The performance differences between these methodologies are substantial. Simulation studies demonstrate that the NCGC method is extremely conservative with very small power, while the Parham method is very liberal with high false discovery rates. In contrast, the PTE-based methodology achieves better control of FDR while maintaining good power [40]. This balance is particularly important when validating enzyme functions, where both false positives (misinterpreting non-specific activity as true function) and false negatives (failing to detect true activity) can perpetuate annotation errors.
Universal versus Coupled Assay Platforms
A critical decision in assay development is selecting between universal assay platforms that detect common enzymatic products versus coupled assay systems that use secondary enzymes to generate detectable signals. Universal assays like Transcreener detect products such as ADP (for kinases, ATPases, GTPases) or SAH (for methyltransferases) using competitive immunodetection with various fluorescent formats (FI, FP, TR-FRET) [41]. These platforms offer broad applicability across enzyme classes and simplified "mix-and-read" formats that reduce variability and increase throughput.
Coupled assays, while historically important and well-established, introduce additional variables through the coupling enzymes and may be susceptible to interference from compounds that affect either the primary or coupling enzymes. This is particularly problematic when screening enzymes with uncertain annotations, as the additional complexity makes results more difficult to interpret. Universal assays provide a more direct measurement of enzymatic activity, making them preferable for functional validation studies.
Essential Research Tools for qHTS Assay Development
Successful implementation of qHTS requires specialized reagents, tools, and platforms. The following table summarizes key solutions for robust assay development and screening.
Table 3: Research Reagent Solutions for qHTS Assay Development
| Tool/Reagent | Function | Application Notes |
|---|---|---|
| Transcreener Platform | Universal assay technology detecting ADP, SAH, and other common enzymatic products | Broad applicability across enzyme classes; multiple fluorescent formats (FI, FP, TR-FRET) [41] |
| AptaFluor SAH Assay | Aptamer-based TR-FRET detection of S-adenosylhomocysteine for methyltransferases | Homogeneous "mix-and-read" format; no antibodies required [41] |
| Homogeneous "Mix-and-Read" Assays | Detection without separation steps or washing | Simplified automation; reduced variability; ideal for HTS [41] |
| Universal Activity Assays | Detect products of enzymatic reactions common between targets | Enables screening multiple targets within enzyme family with same assay [41] |
| Hill Model Regression Analysis | Nonlinear regression for dose-response curve fitting | Essential for quantifying potency (ED50) and efficacy (θ1) [40] |
| Preliminary Test Estimation (PTE) | Robust statistical analysis accounting for heteroscedasticity and outliers | Controls false discovery rates while maintaining power in qHTS [40] |
| Z'-factor Statistical Metric | Quantitative measure of assay quality and robustness | Z' > 0.5 indicates excellent assay suitable for HTS [41] |
Visualization of qHTS Workflows and Data Analysis
Effective visualization of experimental workflows and data analysis pathways is essential for implementing robust qHTS. The following diagrams illustrate key processes in assay development and data interpretation.
qHTS Assay Development and Validation Workflow
qHTS Assay Development Workflow illustrates the systematic process for developing robust assays, from initial objective definition through final implementation, with emphasis on critical validation steps.
Statistical Analysis Pathway for qHTS Data
qHTS Data Analysis Pathway shows the statistical processing of qHTS data from raw responses through final compound classification, highlighting methodology comparisons that impact false discovery rates.
The integration of robust qHTS methodologies into enzyme function validation represents a critical safeguard against the pervasive problem of database misannotation. With studies indicating that nearly 18% of all sequences in the BRENDA database are annotated to enzyme classes while sharing no similarity or domain architecture to experimentally characterized representatives [1], the need for experimental verification through well-designed assays has never been more pressing. The combination of universal assay platforms that provide broad functional coverage, rigorous statistical approaches that control false discovery rates, and systematic validation protocols creates a framework for generating reliable data that can confidently confirm or refute computational predictions.
As high-throughput technologies continue to evolve, the marriage of careful experimental design with robust analytical methods will remain essential for advancing our understanding of enzyme function. By implementing the best practices outlined in this guide—from initial assay design through final statistical analysis—researchers can ensure that their qHTS campaigns produce data of sufficient quality to make meaningful contributions to functional annotation and drug discovery. In an era of increasingly automated functional prediction, the role of carefully validated experimental data as the ultimate arbiter of enzyme function has never been more important.
The reliable production of soluble, active enzymes is a cornerstone of biochemical research and biopharmaceutical development. However, transitioning from a computational annotation or a genetic sequence to a successfully expressed and folded protein remains a significant challenge. This guide examines the primary hurdles in recombinant protein expression, drawing on data from large-scale experimental trials. It provides a structured comparison of solutions, detailing their experimental basis and efficacy to help researchers select the optimal strategies for their projects, all within the critical context of validating computationally generated enzyme sequences.
The Core Challenge: Bridging the Computational-Experimental Gap
The rapid expansion of computational protein sequence generation has outpaced the capacity for experimental validation. A pivotal study evaluating enzymes generated by neural networks reported that initial, naive generation resulted in mostly inactive sequences, with only 19% of tested sequences (including natural controls) showing activity in vitro [5]. This highlights a critical bottleneck: successful in silico design does not guarantee soluble expression or function in the laboratory. Furthermore, the reliability of starting sequences is a concern; one investigation into a specific enzyme class (EC 1.1.3.15) found that at least 78% of sequences were misannotated in databases, underscoring the importance of experimental verification [1].
Common failure modes include protein misfolding, inclusion body formation, proteolytic degradation, and low yield, often exacerbated by factors like codon bias, toxic effects on the host, and improper handling of signal peptides or multimeric interfaces [5] [42] [43].
Quantitative Insights from Large-Scale Studies
Large-scale expression pipelines provide invaluable data on factors influencing success. Analysis of thousands of protein expression experiments in E. coli has revealed strong correlations between specific sequence features and experimental outcomes [44].
Table 1: Amino Acid Correlations with Expression and Solubility
| Amino Acid / Property | Correlation with Expression | Correlation with Solubility | Notes |
|---|---|---|---|
| Leucine (Leu) | Strong Negative | Strong Negative | Strongest negative correlation among amino acids [44] |
| Isoleucine (Ile) | Slightly Positive | Slightly Positive | Contrasting effect despite similarity to Leu [44] |
| Arginine (Arg) | Negative | Negative | Effect only partially attributable to rare codons [44] |
| Overall Hydrophobicity | Negative | Negative | Effect derives primarily from charged amino acids [44] |
| Charged Residues | Positive (Generally) | Positive (Generally) | Positively charged residues may reduce translation efficiency [44] |
A Strategic Framework for Optimization
Overcoming expression hurdles requires a systematic approach targeting the vector, the host, and the growth environment. The following workflow outlines a proven strategy for optimizing protein expression and solubility.
Experimental Protocols for Key Optimization Steps
1. Small-Scale Expression Time Course This protocol is fundamental for establishing a baseline and optimizing conditions [45] [46].
- Inoculation: Pick a fresh, single colony from a transformed plate and inoculate a small volume of media (e.g., 5 mL LB). Grow overnight at 37°C with shaking.
- Dilution: Dilute the overnight culture 1:100 into fresh media (e.g., 50 mL in a baffled flask for better aeration).
- Growth and Induction: Grow the culture at 37°C with shaking until mid-log phase (OD600 ~0.6-0.9). Take a 1 mL pre-induction sample. Add inducer (e.g., IPTG).
- Induction and Sampling: Induce expression for several hours (e.g., overnight), taking 1 mL samples every hour post-induction.
- Analysis: Pellet the cells from each sample, lyse them (e.g., with lysozyme or sonication), and separate soluble and insoluble fractions by centrifugation. Analyze all samples by SDS-PAGE to assess total expression and solubility over time.
2. Solubility Enhancement via Low-Temperature Induction
- Procedure: After growing the main culture to mid-log phase at 37°C, reduce the incubation temperature to 18°C [42]. Allow the culture to equilibrate for 30-60 minutes before adding inducer. Continue incubation with shaking at 18°C for 16-20 hours (overnight) [42].
- Rationale: Slower protein synthesis at lower temperatures gives the cellular folding machinery more time to process the polypeptide chain, reducing misfolding and aggregation into inclusion bodies.
3. Controlling Basal Expression in T7 Systems
- Using Inhibitor Strains: To combat "leaky" expression, use host strains that co-express T7 Lysozyme (e.g., T7 Express lysY, or strains containing pLysS/pLysE plasmids) [43]. T7 Lysozyme inhibits T7 RNA polymerase, providing tighter control.
- Using Repressor Strains: Employ strains with enhanced Lac repressor production (e.g., containing the
lacIqallele) to more effectively block transcription from lac-based promoters before induction [43]. - Glucose Supplementation: Adding 1% glucose to the growth medium can suppress basal expression from the lacUV5 promoter in DE3 strains by reducing cAMP levels [43].
Comparative Analysis of Expression Solutions
Different challenges require tailored solutions. The table below compares the performance and application of common strategies.
Table 2: Comparison of Key Expression Optimization Strategies
| Strategy | Mechanism of Action | Typical Application | Experimental Evidence/Notes |
|---|---|---|---|
| Fusion Tags (e.g., MBP) | Enhances solubility; simplifies purification | Proteins prone to aggregation | pMAL system: MBP fusion allows purification via amylose column; can be cleaved off [43]. |
| Low-Temp Induction | Slows synthesis, improves folding | Proteins forming inclusion bodies at 37°C | Consensus protocol: Induction at 18°C overnight drastically improves soluble yield [42]. |
| Rare tRNA Strains | Supplies tRNAs for rare codons; prevents stalling | Genes with codons rare in E. coli (e.g., Arg, Ile) | Use of BL21(DE3)-RIL cells improved expression despite plasmid-encoded tRNAs [44] [42]. |
| Tuned Expression (e.g., Lemo21) | Precisely controls expression level via rhamnose | Toxic proteins; proteins prone to insolubility | Yields soluble protein by keeping expression below host toxicity/aggregation threshold [43]. |
| Chaperone Co-expression | Assists in vivo protein folding | Complex multidomain proteins | Co-expression of GroEL, DnaK, or ClpB can improve solubility of some targets [42] [43]. |
| Disulfide Bond Strains (SHuffle) | Allows correct disulfide formation in cytoplasm | Proteins requiring disulfide bonds for stability | Cytoplasmic DsbC expression in an oxidizing background enables proper folding [43]. |
The Scientist's Toolkit: Essential Research Reagents
| Reagent / Material | Function | Example Products / Strains |
|---|---|---|
| Tightly-Regulated Vectors | Controls basal ("leaky") expression to avoid host toxicity. | pET (T7 lacO), pMAL [42] [43] |
| Specialized Host Strains | Provides specific cellular environments for different protein challenges. | BL21(DE3) (standard), T7 Express lysY / pLysS (low leak), BL21(DE3)-RIL (rare tRNAs), SHuffle (disulfide bonds) [42] [43] |
| Solubility Enhancement Tags | Fuses to target protein to improve folding and solubility; aids purification. | Maltose-Binding Protein (MBP), Glutathione-S-transferase (GST) [43] |
| Protease Inhibitors | Prevents degradation of the target protein during cell lysis and purification. | Commercial protease inhibitor cocktails [43] |
| Tunable Inducers | Allows fine control over expression levels to find the optimal balance between yield and solubility. | L-rhamnose (for PrhaBAD promoter) [43] |
Navigating protein expression and solubility challenges is a multifaceted process that benefits immensely from a data-driven and systematic approach. Large-scale studies have illuminated key sequence-level predictors of success, while robust experimental frameworks provide a clear path for optimization. By strategically selecting from a well-stocked toolkit of vectors, host strains, and growth conditions—such as employing low-temperature induction, solubility tags, and tightly controlled expression systems—researchers can significantly increase their chances of producing soluble, active enzyme variants. This is especially critical in the modern research landscape, where the ability to experimentally validate the ever-growing number of computationally generated and annotated sequences is paramount to advancing both basic science and drug development.
In the field of enzyme engineering, a significant challenge lies in efficiently identifying functional proteins from the vast number of variants generated by computational models. While generative AI can produce thousands of novel enzyme sequences, a large proportion often fails to express, fold, or function in the laboratory. This article examines the Composite Metrics for Protein Sequence Selection (COMPSS) framework, a computational filter designed to address this bottleneck by significantly improving the rate of experimental success in identifying active enzymes. Framed within the broader thesis of validating enzyme function from computational annotations, we will objectively compare the performance of COMPSS against other established generative models and provide the detailed experimental data and protocols that underpin these findings.
The Need for Robust Computational Filters in Enzyme Validation
Generative protein sequence models, such as those based on generative adversarial networks (GANs), ancestral sequence reconstruction (ASR), and protein language models, have revolutionized enzyme engineering by sampling novel sequences beyond natural diversity [5]. However, their initial promise was hampered by a high experimental failure rate. Early, "naive" generation from these models resulted in sequences where as few as 19% were experimentally active, including natural test sequences [5]. This inefficiency stems from various factors, including mutations that disrupt folding, poor stability, and issues with heterologous expression in workhorse organisms like E. coli.
This high attrition rate underscores the critical need for effective computational filters. Without them, researchers face costly and time-consuming experimental workflows cluttered with non-functional variants. The transition from mere sequence generation to reliable function prediction represents the next frontier in computational enzyme design.
Comparative Performance: COMPSS vs. Alternative Generative Models
The COMPSS framework was developed through a rigorous, multi-round experimental process. Researchers generated over 30,000 sequences from three contrasting generative models—ProteinGAN (a GAN), ESM-MSA (a transformer-based language model), and ASR (a phylogeny-based statistical model)—focused on two enzyme families: malate dehydrogenase (MDH) and copper superoxide dismutase (CuSOD) [5]. By expressing and purifying over 500 natural and generated sequences, they benchmarked 20 diverse computational metrics to create a composite filter that predicts in vitro enzyme activity.
The table below summarizes the key experimental success rates that demonstrate the superiority of the COMPSS-filtered approach.
Table 1: Experimental Success Rates of Generative Models
| Generative Model | Description | Round 1: Naive Generation Success Rate | Success Rate with COMPSS Filter |
|---|---|---|---|
| Ancestral Sequence Reconstruction (ASR) | A phylogeny-based statistical model [5]. | CuSOD: 50% (9/18)\nMDH: 56% (10/18) [5] | Improved rate of experimental success by 50–150% [5] |
| ProteinGAN | A convolutional neural network with attention trained as a GAN [5]. | CuSOD: ~11% (2/18)\nMDH: 0% (0/18) [5] | Improved rate of experimental success by 50–150% [5] |
| ESM-MSA | A transformer-based multiple-sequence alignment language model [5]. | CuSOD: 0% (0/18)\nMDH: 0% (0/18) [5] | Improved rate of experimental success by 50–150% [5] |
| Overall (Including Natural) | Combined performance before optimization [5]. | 19% active [5] | Composite metric allows selection of up to 100% of phylogenetically diverse functional sequences [5] |
The data shows that while ASR initially produced more stable and functional variants, the application of the COMPSS filter dramatically enhanced the success rate across all model types. The framework is noted for its generalizability to any protein family.
Inside the COMPSS Framework: Metrics and Methodology
The power of COMPSS lies in its integration of three distinct classes of computational metrics, moving beyond reliance on any single score.
Core Computational Metrics
COMPSS synthesizes three types of metrics for a robust evaluation [5]:
- Alignment-based metrics: Such as sequence identity to the closest natural sequence or BLOSUM62 scores. These are effective for detecting general sequence properties but can miss complex epistatic interactions [5].
- Alignment-free metrics: Often based on the likelihoods computed by protein language models. These are fast to compute and can identify sequence defects without requiring homology searches [5].
- Structure-based metrics: Including Rosetta-based scores and AlphaFold2 residue confidence scores. These use predicted atomic coordinates to assess function but can be computationally expensive for large-scale screening [5].
Key Experimental Protocol
The experimental validation of COMPSS involved a standardized workflow to assess whether a generated sequence could be expressed, folded, and function in a biologically relevant context [5].
Detailed Methodology:
- Sequence Generation & Selection: Models were trained on curated sets of CuSOD (6,003 sequences) and MDH (4,765 sequences) from UniProt. Over 30,000 novel sequences were generated [5].
- Construct Design: Sequences were truncated to their core Pfam domains to remove extraneous signal peptides or transmembrane domains that could hinder heterologous expression in E. coli [5].
- Protein Expression & Purification: Over 500 selected natural and generated sequences were expressed and purified from E. coli [5].
- Activity Assay: Function was assessed via spectrophotometric enzyme activity assays. A variant was deemed "experimentally successful" if its in vitro activity was measurably above background levels [5].
The diagram below illustrates the iterative development and validation workflow of the COMPSS framework.
The Scientist's Toolkit: Research Reagent Solutions
Implementing a workflow like COMPSS requires a suite of specific biological and computational resources. The following table details the key reagents and tools essential for this research.
Table 2: Essential Research Reagents and Tools
| Item Name | Function / Application in the Workflow |
|---|---|
| Malate Dehydrogenase (MDH) & Copper Superoxide Dismutase (CuSOD) | Model enzyme families for benchmarking generative models and computational metrics [5]. |
| Escherichia coli (E. coli) Expression System | A heterologous host for the expression and purification of generated protein variants [5]. |
| Spectrophotometric Activity Assays | In vitro method to quantitatively measure enzyme function and define experimental success above background [5]. |
| Pfam Database | A curated resource of protein families and domains, used to define and truncate sequence constructs for testing [5]. |
| UniProt Database | A comprehensive repository of protein sequences, used to curate training sets for the generative models [5]. |
| Google Colab Notebooks | Provided by the COMPSS developers to facilitate the general application of the framework to other protein families [5]. |
The transition from generating novel enzyme sequences to reliably predicting their function is a central challenge in computational biology. The experimental data demonstrates that the COMPSS framework represents a significant leap forward, transforming generative models from sources of largely inactive sequences into powerful tools for engineering diverse, functional enzymes. By implementing a composite filter that intelligently combines multiple computational metrics, COMPSS boosts experimental success rates by 50% to 150%, providing researchers and drug development professionals with a validated, generalizable method to prioritize active variants and accelerate discovery.
Validating the function of enzymes derived from computational predictions presents a significant challenge in modern bioengineering. A substantial gap often exists between in silico annotations and experimentally confirmed activity, with misannotation rates in some enzyme classes exceeding 78% [9]. This discrepancy frequently stems from overlooking critical protein features that impact experimental outcomes. Signal peptides, inappropriate truncations, and multimeric states constitute three fundamental pitfalls that can compromise enzyme activity despite favorable computational scores. This guide examines these pitfalls through experimental data and provides methodological frameworks for researchers to enhance the accuracy of enzyme function validation, ultimately bridging the gap between computational prediction and experimental reality in drug development and enzyme engineering.
Pitfall 1: Signal Peptides and Protein Localization
Structural and Functional Mechanisms
Signal peptides (SPs) are short amino acid sequences (typically 15-30 residues) located at the N-terminus of nascent proteins that direct cellular localization and translocation [47]. These peptides follow a characteristic three-region structure consisting of a positively charged N-region, a hydrophobic H-region, and a polar C-region with the signal peptidase cleavage site [47]. The N-region's positive charges facilitate interaction with signal recognition particles (SRPs) and the Sec translocation machinery, initiating the transmembrane transport process [47].
In recombinant expression systems, failure to properly account for signal peptides can severely impact experimental outcomes. Bacterial signal peptides typically direct proteins to the periplasmic space or extracellular environment, while eukaryotic signals target the endoplasmic reticulum [47]. Retaining native signal peptides in heterologous systems often prevents proper localization, while inappropriate removal can disrupt protein folding and function.
Experimental Evidence and Impact
Research demonstrates that signal peptide mishandling significantly contributes to experimental failure. In a comprehensive study evaluating computationally generated enzymes, natural test sequences with predicted signal peptides or transmembrane domains were significantly overrepresented in the non-active set (one-tailed Fisher test, P = 0.046) [5]. The strategic removal of signal peptides at their predicted cleavage sites dramatically improved functional outcomes. For bacterial copper superoxide dismutase (CuSOD) proteins, which typically utilize secretion signals, truncation at predicted cleavage sites restored activity in 8 of 14 previously inactive sequences [5].
Table 1: Impact of Signal Peptide Management on Experimental Outcomes
| Protein Type | Signal Peptide Handling | Expression Success | Functional Activity |
|---|---|---|---|
| Bacterial CuSOD | Retained native signal peptide | Poor | Inactive |
| Bacterial CuSOD | Truncated at cleavage site | Improved | 57% (8/14) active |
| Eukaryotic CuSOD | Native sequence (no signal) | Good | Active |
| General Recombinant Proteins | Rational SP optimization | 1.6-3x improvement | Varies by system |
Recommended Experimental Protocol
- Computational Prediction: Utilize SignalP 6.0 or similar tools incorporating deep learning models to identify signal peptides and their cleavage sites [47].
- Cleavage Site Verification: Confirm predicted cleavage sites using multiple algorithms to increase confidence.
- Construct Design: For heterologous expression, remove signal peptides precisely at cleavage sites or select appropriate signal peptides matched to the expression system.
- Localization Validation: Employ cellular fractionation or microscopy to confirm proper protein localization.
- Functional Assessment: Compare activity between constructs with and without signal peptide management.
Pitfall 2: Terminal Truncations and Domain Integrity
The Truncation Balance
Protein truncations represent a double-edged sword in enzyme engineering. While strategic truncation of disordered regions can enhance expression and activity, improper truncation can disrupt essential structural elements and functional domains. The key challenge lies in distinguishing between beneficial removal of flexible termini and detrimental excision of structurally or functionally critical regions.
Experimental Evidence of Truncation Effects
Disorder prediction-based truncation of Bacillus naganoensis pullulanase (PUL) demonstrates how strategic truncation impacts enzyme performance [48]. Systematic removal of N-terminal disordered regions yielded varied outcomes:
Table 2: Impact of N-terminal Truncations on Pullulanase Function
| Construct | Truncation Length | Protein Production | Specific Activity | Catalytic Efficiency |
|---|---|---|---|---|
| PULΔN5 | 5 residues | Increased | 324 U/mg (1.18x) | Enhanced |
| PULΔN22 | 22 residues | Decreased | Reduced | Impaired |
| PULΔN45 | 45 residues | Similar to wild-type | Similar | Enhanced |
| PULΔN106 | 106 residues | Increased | 382 U/mg (1.38x) | Enhanced |
The most successful truncations (PULΔN5 and PULΔN106) targeted regions with high disorder propensity while preserving catalytic domains [48]. These constructs exhibited both improved expression levels and enhanced specific activity, indicating proper folding and functional optimization.
Conversely, inappropriate truncation presents significant risks. In the evaluation of generated malate dehydrogenase (MDH) and CuSOD sequences, overtruncation emerged as a major cause of experimental failure [5]. For CuSOD, truncations often removed residues critical for dimer interface formation, thereby disrupting the quaternary structure necessary for function. Equivalent truncations applied to positive-control enzymes (human SOD1 and Potentilla atrosanguinea CuSOD) confirmed complete loss of activity, validating that the truncations rather than sequence defects caused functional impairment [5].
Recommended Experimental Protocol
- Disorder Prediction: Utilize meta-servers like Disorder Prediction Meta-Server (DisMeta) combining multiple algorithms (DISEMBL, DISOPRED2, IUPred, etc.) [48].
- Domain Mapping: Identify functional domains using Pfam and similar databases to avoid critical region excision.
- Construct Design: Target truncations to regions with high consensus disorder predictions outside functional domains.
- Multi-Construct Approach: Generate multiple truncation variants to empirically determine optimal boundaries.
- Structural Validation: When possible, verify overall structure retention through circular dichroism or similar techniques.
Pitfall 3: Multimeric States and Quaternary Structure
Cooperativity Without Conformational Changes
Many functional enzymes operate as multimeric complexes, with subunit interactions fundamentally influencing catalytic efficiency and regulation. The bacterial heptose isomerase GmhA exemplifies how multimeric organization impacts function without substantial conformational changes [49]. This tetrameric enzyme displays both positive cooperativity (Hill coefficient of 1.5-2) and half-site reactivity, where only two of four potential active sites function simultaneously [49].
Experimental Evidence of Multimeric Effects
GmhA's unique cooperativity arises from a delicate network of hydrogen bonds and a water channel connecting paired active sites, rather than large-scale conformational changes [49]. This sophisticated control mechanism enables the enzyme to precisely regulate catalytic activity through its quaternary structure.
Experimental manipulation of GmhA's multimeric state produced surprising results. When three of four active sites were rendered inoperative through mutation, the enzyme retained approximately 40% of wild-type activity [49]. Even more remarkably, the single-active-site variant maintained cooperative kinetics despite having only one functional site, suggesting that non-catalytic subunits still influence substrate binding and enzyme function.
Table 3: Impact of Active Site Mutations on Tetrameric GmhA Kinetics
| Enzyme Variant | Functional Active Sites | kcat per Protomer (s⁻¹) | kcat per Intact Site (s⁻¹) | Cooperativity |
|---|---|---|---|---|
| Wild-type | 4 | 0.44 ± 0.02 | 0.44 ± 0.02 | Positive (Hill 1.5-2) |
| Triple mutant | 1 | 0.144 ± 0.008 | 0.57 ± 0.03 | Positive cooperativity maintained |
| Single mutant | 3 | 0.43 ± 0.02 | 0.57 ± 0.02 | Positive cooperativity maintained |
Advanced techniques like time-resolved electrospray ionization mass spectrometry (ESI-MS) enable direct observation of multimeric enzyme catalysis. This methodology successfully detected intact tetrameric complexes of 4-hydroxybenzoyl-coenzyme A thioesterase with up to four natural substrate molecules bound, revealing real-time substrate and product occupancy during the catalytic cycle [50].
Recommended Experimental Protocol
- Quaternary Structure Prediction: Utilize PISA or similar tools to model multimeric interfaces.
- Size-Exclusion Chromatography: Determine native molecular weight and oligomeric state.
- Cross-linking Studies: Confirm subunit interactions under native conditions.
- Kinetic Analysis: Measure enzyme kinetics across substrate concentrations to detect cooperativity.
- Interface Mapping: Identify critical residues at subunit interfaces for targeted mutagenesis.
Integrated Experimental Workflow
Diagram 1: Integrated workflow for validating enzyme function from computational predictions, incorporating analysis of three common pitfalls.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagent Solutions for Enzyme Validation Studies
| Reagent / Tool | Primary Function | Application Notes |
|---|---|---|
| SignalP 6.0 | Signal peptide prediction | Combines BERT language models with conditional random fields for enhanced accuracy [47] |
| Disorder Prediction Meta-Server | Disordered region identification | Integrates eight disorder prediction algorithms for consensus analysis [48] |
| Pfam Database | Protein domain family annotation | Critical for identifying functional domains to avoid detrimental truncations [9] |
| Amplex Red Peroxidase Assay | Hydrogen peroxide detection | Indirect measurement of oxidase activity for high-throughput screening [9] |
| Size-Exclusion Chromatography | Quaternary structure analysis | Determines native oligomeric state and complex stability |
| Rapid-Mixing ESI-MS | Time-resolved kinetics | Monitors substrate binding and product release in multimeric enzymes [50] |
| pET-28a-PelB Vector | Recombinant protein expression | Facilitates secretory expression with removable signal peptide [48] |
Successfully bridging the gap between computationally predicted and experimentally validated enzyme function requires meticulous attention to structural and cellular contexts. Signal peptides must be strategically managed through precise cleavage or system-matched selection. Terminal truncations should leverage disorder predictions while respecting domain integrity. Multimeric states demand preservation of quaternary structures and recognition of cooperative effects. By integrating computational predictions with rigorous experimental validation protocols that address these pitfalls, researchers can significantly improve the success rate of enzyme engineering projects. The provided frameworks and methodologies offer practical pathways for more reliable translation of in silico designs to functionally confirmed enzymes, accelerating progress in therapeutic development and industrial biocatalysis.
Benchmarking Truth: Establishing Standards for Model Performance and Validation
The accurate prediction of enzyme function from sequence and structural data is a cornerstone of modern bioinformatics and drug discovery. As the volume of genetically sequenced data vastly outpaces the capacity for experimental characterization, researchers increasingly rely on computational scores to annotate function and select promising candidates for further study. However, the relationship between these computational predictions and actual experimental activity is complex and requires rigorous validation. This guide objectively compares the performance of various computational evaluation methods against experimental results, providing a framework for researchers to assess the predictive power of different metrics in the context of validating enzyme function.
Experimental Protocols in Enzyme Validation
To understand the performance of computational metrics, it is essential to examine the experimental methodologies used to generate validation data. The following protocols represent key approaches cited in recent literature for experimentally determining enzyme activity.
High-Throughput Screening for Enzyme Annotation Validation
A comprehensive study investigating misannotation in the S-2-hydroxyacid oxidase enzyme class (EC 1.1.3.15) employed a high-throughput experimental platform to validate computational predictions [9]. The methodology proceeded as follows:
- Gene Selection and Synthesis: 122 representative sequences spanning the diversity of EC 1.1.3.15 were selected based on computational analysis of the BRENDA database. These genes were synthesized for experimental testing [9].
- Recombinant Expression: The synthesized genes were cloned and expressed recombinantly in Escherichia coli using a high-throughput setup. This step assessed whether the proteins could be produced in a soluble, likely functional form [9].
- Solubility Assessment: After expression, researchers determined that 65 of the 122 proteins (53%) were in a soluble state, with archaeal and eukaryotic proteins showing proportionally lower solubility than bacterial proteins [9].
- Activity Assay: The soluble proteins were tested for S-2-hydroxy acid oxidase activity using an Amplex Red peroxide detection assay. This spectrophotometric method detects hydrogen peroxide, a byproduct of the oxidase reaction, providing a quantitative measure of enzymatic activity [9].
Multi-Round Benchmarking of Generative Models
A separate study focusing on malate dehydrogenase (MDH) and copper superoxide dismutase (CuSOD) established a rigorous benchmark for computational metrics over three rounds of experimentation [5]:
- Sequence Generation: Researchers generated over 30,000 sequences using three contrasting generative models: Ancestral Sequence Reconstruction (ASR), a Generative Adversarial Network (ProteinGAN), and a protein language model (ESM-MSA) [5].
- Sequence Selection and Purification: For experimental testing, they selected 144 generated sequences plus natural test sequences, all with 70-80% identity to the closest natural training sequence. These sequences were expressed and purified [5].
- Activity Determination: A protein was considered "experimentally successful" only if it could be expressed and folded in E. coli and demonstrated activity above background levels in a standardized in vitro assay. For CuSOD, this involved spectrophotometric analysis of superoxide dismutation [5].
- Iterative Refinement: Initial experiments revealed issues such over-truncation that removed critical structural elements. The experimental design was subsequently refined, and additional sequences were tested to control for these factors. This iterative process informed the development of better computational filters [5].
Comparative Performance of Computational Metrics
The critical challenge in computational enzymology is selecting the right metrics to predict which sequences will show experimental activity. The following analysis compares the performance of various computational approaches based on experimental validation studies.
Performance of Generative Models
The ability of different generative models to produce functional enzymes varies significantly. The table below summarizes the experimental success rates for two enzyme families as reported in a benchmark study [5].
Table 1: Experimental Success Rates of Generative Models
| Generative Model | CuSOD Active/Tested | CuSOD Success Rate | MDH Active/Tested | MDH Success Rate |
|---|---|---|---|---|
| Ancestral Sequence Reconstruction (ASR) | 9/18 | 50.0% | 10/18 | 55.6% |
| Generative Adversarial Network (ProteinGAN) | 2/18 | 11.1% | 0/18 | 0.0% |
| Protein Language Model (ESM-MSA) | 0/18 | 0.0% | 0/18 | 0.0% |
| Natural Test Sequences | 6/~14* | ~42.9%* | 6/18 | 33.3% |
Note: *The exact number of natural CuSOD sequences tested in the first round is not explicitly stated; the value reflects active sequences from a subsequent pre-test group [5].
The data reveals that ASR significantly outperformed other generative methods, producing active enzymes at a rate comparable to or even exceeding that of natural test sequences. In contrast, the deep learning models (ProteinGAN and ESM-MSA) initially failed to generate any active MDHs and only a few active CuSODs [5].
Performance of Computational Filtering Metrics
To improve the success rate of generated sequences, researchers developed and tested a composite filter called COMPSS (Composite Metrics for Protein Sequence Selection). The table below summarizes the improvement achieved by applying computational filters to select sequences for testing [5].
Table 2: Efficacy of Computational Filtering
| Filtering Strategy | Improvement in Experimental Success Rate | Key Metrics Included |
|---|---|---|
| Naive Selection (No Filter) | Baseline | Sequence identity, BLOSUM62 score |
| COMPSS Filter | 50% - 150% improvement | Combination of alignment-based, alignment-free, and structure-based metrics |
The COMPSS framework integrated multiple metrics, moving beyond simple alignment-based scores like sequence identity to include alignment-free methods (e.g., likelihoods from protein language models) and structure-based scores (e.g., AlphaFold2 confidence scores and Rosetta energy scores). This multi-faceted approach proved far more effective at identifying functional sequences [5].
The Problem of Database Misannotation
Computational predictions are only as reliable as the data on which they are based. A systematic investigation of the EC 1.1.3.15 enzyme class highlights a critical source of error: widespread database misannotation [9] [39].
Table 3: Scope of Misannotation in EC 1.1.3.15
| Analysis Method | Finding | Implication |
|---|---|---|
| Sequence Identity Analysis | 79% of sequences shared <25% identity to a characterized enzyme [9]. | Automated annotation transfer is highly unreliable for this class. |
| Domain Architecture Analysis | Only 22.5% of sequences contained the canonical FMN-dependent dehydrogenase domain (PF01070) [9]. | Majority of sequences likely have completely different functions. |
| Experimental Validation | At least 78% of sequences in the class were misannotated [9]. | Computational predictions based on these annotations are flawed. |
| Database-Wide Analysis | ~18% of all sequences in BRENDA are annotated to classes with no similarity to characterized representatives [9]. | Misannotation is a pervasive problem across enzyme classes. |
This study demonstrates that a significant proportion of functional annotations in public databases are erroneous, which perpetuates errors through automated annotation pipelines and confounds the training and evaluation of computational models [9].
Visualization of Experimental and Computational Workflows
The following diagrams illustrate the key workflows for the experimental validation of computational predictions and the structure of a successful computational filtering framework.
Workflow for Validating Computational Predictions
COMPSS: A Multi-Metric Filtering Framework
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and materials essential for conducting the experimental validation protocols described in this guide.
Table 4: Key Research Reagents for Enzyme Validation
| Reagent/Material | Function in Experimental Protocol |
|---|---|
| Amplex Red Peroxide Assay Kit | Fluorometric detection of hydrogen peroxide produced by oxidase enzymes; used for high-throughput activity screening [9]. |
| Heterologous Expression System (E. coli) | Workhorse for recombinant protein production; allows for scalable expression of target enzyme sequences [5] [9]. |
| Chromatography Purification Systems | Purification of expressed enzymes (e.g., affinity, size-exclusion) to obtain clean protein for functional assays [5]. |
| Synthesized Gene Sequences | Source material for expression; represents the computational predictions being tested [5] [9]. |
| Cell Culture Reagents | Media, antibiotics, and inducers (e.g., IPTG) for growing expression cultures and triggering protein production [5] [9]. |
| Spectrophotometer / Plate Reader | Essential instrument for measuring enzyme activity through absorbance or fluorescence in kinetic assays [5] [9]. |
| 3D Bioprinting Hydrogels (e.g., PEG-based) | Advanced cell culture matrix for creating more physiologically relevant 3D models for functional testing [51]. |
The comparative analysis of computational scores and experimental activity reveals a nuanced landscape. No single computational metric is sufficient to reliably predict enzyme function. While generative models like ASR show promise, the high failure rates of other advanced models underscore the complexity of the sequence-function relationship. The most successful strategy involves a multi-faceted approach, combining diverse computational metrics into composite filters like COMPSS, which can significantly improve experimental hit rates. Furthermore, researchers must be cognizant of the pervasive issue of database misannotation, which can fundamentally undermine both training and evaluation of predictive models. As the field advances, the continued iterative cycle of computational prediction and rigorous experimental validation, using the standardized protocols and reagents outlined here, remains essential for progress.
This guide provides an objective performance comparison of the EZSpecificity artificial intelligence (AI) tool against a prior state-of-the-art model for predicting enzyme-substrate specificity, with a focused analysis on halogenase enzymes. The evaluation is situated within the broader research context of validating computational annotations of enzyme function, a critical step for reliable biocatalyst design. For researchers in drug development and synthetic biology, the transition from in silico annotation to experimentally validated function remains a significant challenge. This case study demonstrates that EZSpecificity, a model leveraging a cross-attention graph neural network architecture, achieves a 91.7% accuracy in identifying reactive halogenase substrates, substantially outperforming the existing Enzyme Substrate Prediction (ESP) model, which showed 58.3% accuracy in the same experimental validation [12] [52] [53]. The following sections detail the experimental protocols, quantitative results, and key resources that underpin this performance leap.
Experimental Setup & Methodologies
A consistent, rigorous methodology was applied to benchmark EZSpecificity against the leading model, ESP, ensuring a fair comparison relevant to real-world enzyme discovery pipelines.
Model Architectures
- EZSpecificity: This model employs a cross-attention-empowered SE(3)-equivariant graph neural network [12]. This architecture is specifically designed to process the three-dimensional (3D) structure of an enzyme's active site and the complicated transition state of the reaction. The SE(3)-equivariance property ensures that model predictions are consistent with rotations and translations of the input molecular structures, a crucial feature for robust biochemical prediction. The cross-attention mechanism allows the model to dynamically focus on the most relevant parts of the enzyme and substrate during their interaction simulation [12].
- ESP (Enzyme Substrate Prediction): As the previous state-of-the-art model used for comparison, ESP's specific architecture is not detailed in the provided search results. Its performance serves as the baseline against which EZSpecificity's advancements are measured [12] [54].
Benchmarking Dataset and Validation
The comparative study involved a four-scenario test designed to mimic real-world applications, followed by a definitive experimental validation [52] [55].
- Enzyme Class: The validation focused on halogenase enzymes, which are increasingly used in the synthesis of bioactive molecules but are often poorly characterized [12] [54].
- Experimental Scale: The key validation experiment tested 8 halogenase enzymes against a panel of 78 substrates [12] [55].
- Data Foundation: A critical innovation behind EZSpecificity is its training on a comprehensive, tailor-made database of enzyme-substrate interactions. This was expanded beyond existing experimental data through millions of molecular docking calculations performed by Shukla's group. These simulations zoomed in on atomic-level interactions between enzymes and substrates, providing a rich dataset on how enzymes of various classes conform around different substrates [52] [53] [55].
- Performance Metric: The primary metric for the halogenase validation was prediction accuracy, defined as the model's ability to correctly identify the single potential reactive substrate for a given enzyme [12].
The workflow below illustrates the integrated computational and experimental process for developing and validating EZSpecificity.
Performance Data & Comparative Analysis
The following table summarizes the quantitative results from the head-to-head comparison between EZSpecificity and ESP, particularly in the challenging task of predicting halogenase substrate specificity.
Table 1: Comparative Performance of EZSpecificity vs. ESP on Halogenase Substrate Prediction
| Model | Underlying Architecture | Key Innovation | Test Scenario | Reported Accuracy |
|---|---|---|---|---|
| EZSpecificity | Cross-attention SE(3)-equivariant Graph Neural Network [12] | Integrates enzyme structure and docking simulation data [52] [55] | Halogenase Validation (8 enzymes, 78 substrates) | 91.7% [12] [54] |
| ESP | Not Specified in Sources | Previous State-of-the-Art | Halogenase Validation (8 enzymes, 78 substrates) | 58.3% [12] [54] |
Analysis of Results
The data indicates a profound improvement in prediction accuracy with EZSpecificity. Its 91.7% accuracy in the halogenase validation experiment represents a 33.4-percentage-point increase over the ESP model. This performance leap is attributed to two main factors:
- Advanced Model Architecture: The use of a structure-aware, equivariant graph neural network allows EZSpecificity to more effectively capture the physical and geometric constraints of the enzyme-substrate binding interaction, moving beyond simpler "lock and key" analogies to model "induced fit" [12] [52] [53].
- Enhanced Data Foundation: The incorporation of millions of dedicated docking simulations addressed a critical gap in publicly available experimental data, providing the model with a much more detailed understanding of atomic-level interactions [52] [55].
The Scientist's Toolkit
The development and validation of EZSpecificity relied on a suite of computational and experimental resources. The table below details key reagents and solutions essential for replicating such a study or applying the tool in a research setting.
Table 2: Key Research Reagent Solutions for Enzyme Specificity Prediction
| Tool / Reagent | Type | Primary Function in Research | Relevance to EZSpecificity |
|---|---|---|---|
| EZSpecificity Tool | Software / AI Model | Predicts substrate specificity from enzyme sequence and substrate structure [52]. | The core technology under evaluation; provides a user-friendly interface for researchers [53] [54]. |
| Molecular Docking Software | Computational Tool | Simulates atomic-level interactions between enzymes and substrates to predict binding affinity and pose [52]. | Used to generate millions of docking calculations that expanded the training dataset, crucial for model accuracy [55]. |
| Halogenase Enzymes | Biological Reagent | A class of enzymes that catalyze the incorporation of halogen atoms into substrates [12]. | Served as the primary test case for experimental validation of predictions [12] [54]. |
| Substrate Library | Chemical Library | A curated collection of diverse small molecules used to probe enzyme activity [12]. | The panel of 78 substrates was used to experimentally challenge the model's predictions [12]. |
| CLEAN AI Model | Software / AI Model | Predicts an enzyme's function from its amino acid sequence [52]. | A complementary tool mentioned by developers, useful for broader enzyme function annotation [52] [53]. |
The validation of enzyme function from computational annotations represents a critical bottleneck in biocatalyst discovery and drug development. This process relies heavily on the accurate interpretation and generation of multi-modal data, including protein sequences, structural information, and functional descriptors. Generative Artificial Intelligence (AI) models have emerged as powerful tools for accelerating this research pipeline by predicting enzyme properties, generating novel protein sequences, and annotating functional characteristics. This guide provides a comprehensive, data-driven comparison of three pivotal classes of generative models—Automatic Speech Recognition (ASR) systems for scientific discourse conversion, Generative Adversarial Networks (GANs) for molecular structure generation, and Large Language Models (LLMs) for biological sequence analysis—to inform their optimal application within enzymology and pharmaceutical research contexts. By evaluating these technologies through standardized experimental frameworks and performance metrics, we aim to equip researchers with the empirical evidence necessary to select appropriate computational tools for validating enzyme function predictions.
Comparative Performance Analysis of Generative Models
Automatic Speech Recognition (ASR) Systems
Automatic Speech Recognition technologies enable the conversion of scientific discourse, laboratory notes, and lecture content into searchable, analyzable text, thereby facilitating knowledge extraction and data curation in research environments. The performance of contemporary ASR systems varies significantly across different operational conditions, with accuracy being paramount for scientific applications where terminological precision is non-negotiable.
Table 1: Performance Comparison of Leading Commercial ASR Models (2025)
| Model | Word Error Rate (WER) | Languages Supported | Latency | Customization | Optimal Research Use Case |
|---|---|---|---|---|---|
| GPT-4o-Transcribe | <5% | 50+ | Ultra-low | Moderate | High-accuracy research apps |
| ElevenLabs Scribe | ~3.3% | 99 | Low | Limited | English-focused documentation |
| Deepgram Nova-3 | <5% | 40+ | ~0.1 RTF | Yes | Real-time scientific streaming |
| Google Speech-to-Text | <7% | 100+ | Low | Yes | Global collaborative projects |
| AssemblyAI Universal-2 | <6% | 100+ | ~270ms | Yes | Enterprise-scale research |
| Azure AI Speech | <8% | 140+ | Low | Yes | Enterprise integration |
Table 2: Performance Comparison of Open-Source ASR Champions (2025)
| Model | Parameters | Word Error Rate (WER) | Real-Time Factor (RTFx) | License | Notable Features |
|---|---|---|---|---|---|
| Canary Qwen 2.5B | 2.5B | 5.63% | 418 | Apache 2.0 | State-of-the-art accuracy, hybrid LLM architecture |
| Granite Speech 3.3 | 8B | 5.85% | 31 | Apache 2.0 | Large multi-language model, translation |
| Parakeet TDT 0.6B | 600M | 6.05% | 3386 | CC-BY-4.0 | Extremely fast batch transcription |
| Whisper Large V3 | 809M | 10-12% | 216 | MIT | Multilingual versatility, strong community |
| Kyutai 2.6B | 2.6B | 6.4% | 88 | CC-BY-4.0 | Optimized for real-time streaming |
The performance differentials illustrated in Tables 1 and 2 highlight critical considerations for research applications. For instance, the exceptionally high Real-Time Factor (RTFx) of Parakeet TDT 0.6B (3386) indicates superior processing efficiency for large-scale audio datasets, such as archived scientific lectures or lengthy laboratory meeting recordings [56]. Conversely, models like Canary Qwen 2.5B achieve best-in-class accuracy (5.63% WER), making them preferable for transcribing critical research discussions or technical terminology where error minimization is essential [57].
Experimental evidence indicates that ASR performance degrades significantly with domain-specific terminology, regional accents, or suboptimal audio conditions, with accuracy drops of 15-30% observed in non-ideal scenarios [57]. This performance characteristic necessitates careful model selection based on specific research environments and audio quality expectations.
Generative Adversarial Networks (GANs)
Generative Adversarial Networks have demonstrated remarkable capabilities in generating molecular structures and simulating compound conformations, providing valuable tools for enzyme substrate prediction and drug candidate design. While comprehensive comparative metrics for specialized scientific GANs are evolving, their architectural innovations continue to address fundamental challenges in molecular generation.
Table 3: GAN Applications in Molecular and Image Synthesis (2025)
| GAN Variant | Training Stability | Mode Coverage | Output Diversity | Notable Research Applications |
|---|---|---|---|---|
| Progressive GANs | Moderate | High | Medium | High-resolution molecular visualization |
| StyleGAN Series | High | High | High | Protein structure generation |
| CycleGAN | Low | Medium | High | Molecular property translation |
| DCGAN | High | Low | Low | Educational molecular modeling |
| WGAN-GP | High | Medium | Medium | 3D conformer generation |
Recent innovations in GAN architectures have focused on improving training stability and output diversity, with 2025 breakthroughs demonstrating enhanced performance in novel image synthesis techniques [58]. These advancements directly benefit molecular visualization and structural prediction tasks in enzymology research, though quantitative performance benchmarks remain highly specific to application domains.
The application of GANs to enzyme research typically involves generating plausible molecular structures that match specific functional annotations or predicting structural modifications that enhance catalytic activity. Successful deployment requires careful consideration of training stability and output diversity to ensure generated structures are both novel and biochemically viable.
Large Language Models
Large Language Models have transcended textual applications to demonstrate remarkable capabilities in biological sequence analysis, protein function prediction, and scientific literature mining. Their performance in research contexts varies considerably based on architecture, training data, and specialization.
Table 4: LLM Performance on Research Prediction Tasks
| Model | Research Outcome Prediction Accuracy | Training Data | Specialized Capabilities |
|---|---|---|---|
| Fine-tuned GPT-4.1 | 77% (test set) | 6,000 idea pairs | Empirical AI research outcome prediction |
| GPT-4.1 (off-the-shelf) | ~50% (random guessing) | General corpus | Broad capabilities, limited research specificity |
| Human NLP Experts | 48.9% | N/A | Domain expertise, contextual understanding |
| IDEATOR (VLM) | 94% ASR (MiniGPT-4 jailbreak) | Multimodal data | Jailbreak generation, security benchmarking |
A landmark 2025 study evaluating LLM capabilities for predicting empirical AI research outcomes demonstrated that a fine-tuned GPT-4.1 system achieved 77% accuracy in predicting which of two research ideas would perform better on benchmark tasks, significantly outperforming both human experts (48.9%) and off-the-shelf frontier models [59]. This capability has profound implications for accelerating enzyme research by prioritizing computational experiments most likely to yield valid functional annotations.
Specialized LLMs and Vision-Language Models (VLMs) have also shown remarkable performance in generating experimental protocols and benchmarking scientific tasks. The IDEATOR system, for instance, achieved a 94% attack success rate in jailbreaking MiniGPT-4 with high transferability to other VLMs [60], highlighting both the capabilities and security considerations of these models in research environments.
Experimental Protocols and Methodologies
ASR Evaluation Framework
The quantitative performance data presented in Section 2.1 derives from standardized evaluation methodologies essential for meaningful model comparison. The benchmark procedures for ASR systems encompass the following critical components:
Audio Dataset Curation: Evaluation corpora must include diverse acoustic conditions mirroring research environments. The 2025 benchmarks incorporated 205 hours of real-world audio from diverse environments including laboratories, offices, and conference settings [61]. Datasets were specifically engineered to include balanced demographic representation across age, gender, and accent profiles, with technical terminology relevant to enzymology and computational biology.
Performance Metrics: Word Error Rate (WER) remains the gold standard metric, calculated as (S + D + I) / N, where S represents substitutions, D deletions, I insertions, and N the total words in the reference transcript. Real-Time Factor (RTF) measures processing efficiency as processing time divided by audio duration, with values below 1.0 indicating faster-than-real-time performance [56]. Latency measurements capture the delay between audio input and text output, critical for interactive research applications.
Domain Adaptation Protocols: Superior performance in research contexts requires model customization through domain-specific vocabulary integration. Best practices include: (1) compiling comprehensive terminology lists of enzyme names, scientific terminology, and technical jargon; (2) implementing custom language model weighting to prioritize scientific terminology; (3) utilizing phrase hints for context-aware recognition during experimental documentation [57].
Diagram 1: ASR Model Evaluation Workflow (Characters: 98)
GAN Training and Validation Protocol
The evaluation of Generative Adversarial Networks for molecular generation requires specialized methodologies to assess both structural validity and functional relevance:
Training Framework: Contemporary GAN training employs the WGAN-GP (Wasserstein GAN with Gradient Penalty) objective function to enhance stability, using the loss function L = E[D(x)] - E[D(G(z))] + λE[(||∇D(εx + (1-ε)G(z))||₂ - 1)²] where D represents the discriminator, G the generator, x real samples, z latent vectors, and ε a random interpolation coefficient [58]. Training incorporates progressive growing techniques where resolution gradually increases from low to high dimensions, particularly beneficial for molecular structure generation.
Evaluation Metrics: The Fréchet Inception Distance (FID) quantifies the similarity between generated and real molecular structures in feature space, with lower values indicating higher quality. The Inception Score (IS) measures both diversity and quality of generated samples. For enzyme-specific applications, domain-specific metrics include: (1) chemical validity rate (percentage of generated structures that are synthetically feasible); (2) functional relevance (docking scores with target enzymes); (3) structural novelty (Tanimoto similarity to known active compounds).
Validation Pipeline: Generated molecular structures undergo multi-stage validation: (1) computational checks for chemical validity and stability; (2) molecular dynamics simulations to assess conformational stability; (3) docking studies with target enzyme structures to predict binding affinity; (4) synthetic accessibility analysis to evaluate laboratory feasibility [58].
LLM Research Outcome Prediction Methodology
The remarkable performance of LLMs in predicting empirical research outcomes, as documented in Section 2.3, stems from rigorously designed experimental protocols:
Benchmark Construction: The test framework comprises 1,585 human-verified idea pairs published after the base model's knowledge cut-off date, ensuring no data contamination [59]. Each example includes: (1) a research goal defined by specific benchmarks with quantitative metrics; (2) two competing ideas with detailed descriptions following standardized formats; (3) binary outcome labels determined by majority voting across multiple benchmarks.
Model Training Protocol: The superior-performing system combines a fine-tuned GPT-4.1 model with an iterative paper retrieval agent. The training process incorporates: (1) pre-training on 6,000 historical idea pairs with outcome labels; (2) iterative retrieval augmentation generating search queries, retrieving relevant papers, summarizing content, and filtering by relevance; (3) multi-round cross-validation with human verification reducing mislabeling rates from 11% to 2.5% through incentivized annotation [59].
Evaluation Methodology: Human expert baselines are established by recruiting 25 domain specialists, with each prediction made by an ensemble of 5 experts spending collectively over 45 minutes per assessment. The system's robustness is validated through stress tests evaluating sensitivity to superficial features like idea complexity and recency, confirming that predictions are based on substantive technical factors rather than confounding variables.
Diagram 2: LLM Research Prediction Methodology (Characters: 99)
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of generative models in enzyme function validation requires both computational resources and experimental materials. The following table details essential components of the integrated research pipeline:
Table 5: Research Reagent Solutions for Enzyme Validation with Generative AI
| Reagent / Resource | Function | Example Specifications |
|---|---|---|
| Specialized ASR Models | Convert scientific discourse to annotated text for knowledge extraction | Canary Qwen 2.5B (5.63% WER, 418 RTFx) |
| Molecular Generation GANs | Produce novel enzyme substrates and inhibitor candidates | StyleGAN variants with chemical validity filters |
| Research Prediction LLMs | Prioritize computational experiments for enzyme function validation | Fine-tuned GPT-4.1 (77% prediction accuracy) |
| Benchmark Datasets | Train and validate model performance on domain-specific tasks | 205 hours real-world audio [61] |
| Multi-Model Fusion Frameworks | Combine outputs from multiple models to enhance accuracy and robustness | Parallel processing architectures [57] |
| Domain Adaptation Tools | Customize general models for enzymology terminology and concepts | Custom vocabulary training, phrase hints |
| Validation Suites | Quantify model performance on enzyme-specific metrics | Functional relevance scores, docking validation |
The toolkit emphasizes integration between computational and experimental approaches, with generative models serving as hypothesis generation engines followed by rigorous empirical validation. This synergistic approach accelerates the enzyme function annotation pipeline while maintaining scientific rigor.
The empirical comparison of generative model performance reveals a complex landscape where no single solution dominates all research scenarios. ASR systems demonstrate mature capabilities for scientific discourse processing, with commercial solutions like ElevenLabs Scribe achieving remarkable accuracy (~3.3% WER) for English-language content while open-source alternatives like Canary Qwen 2.5B offer compelling performance (5.63% WER) with greater customization potential. GAN architectures continue to evolve toward greater stability and diversity in molecular generation, though quantitative benchmarks remain highly application-specific. Most remarkably, LLMs fine-tuned on research outcomes demonstrate unprecedented capabilities in predicting experimental success, significantly outperforming human experts (77% vs. 48.9% accuracy) in forecasting which research ideas will validate successfully [59].
For enzyme function validation research, these findings suggest strategic integration of multiple model classes: employing specialized ASR for knowledge extraction from scientific media, utilizing GANs for molecular structure generation, and leveraging predictive LLMs for prioritizing computational experiments. The multi-model paradigm advocated by leading AI implementers [57] appears particularly relevant to enzymology, where diverse data types and validation frameworks necessitate flexible, robust computational approaches. As generative models continue their rapid advancement, their systematic evaluation through standardized benchmarks and experimental protocols remains essential for realizing their potential in accelerating enzyme discovery and functional characterization.
The exponential growth of genomic data has far outpaced functional annotation, creating a critical bottleneck in biotechnology and drug discovery. While computational methods can often predict general enzyme activity, accurately identifying substrate specificity—particularly for proteins with low sequence identity to characterized homologs—remains a formidable challenge. This guide compares the performance of an integrated structural analysis pipeline against traditional annotation methods, demonstrating through experimental data how evolutionary and structural information combined on a Structural Genomics scale can create motifs that precisely identify enzyme activity and substrate specificity. We provide supporting data and detailed protocols to empower researchers to move beyond catalytic residue identification toward robust, functionally validated active site confirmation.
Elucidating gene function remains a major bottleneck that lags far behind sequence production [62]. The standard approach of searching for functionally characterized sequence homologs becomes unreliable for predicting binding specificity when sequence identity falls below 65-80% [62]. This limitation is particularly problematic for proteins from Structural Genomics initiatives with low sequence identity to other proteins, making functional assignment difficult through traditional means [62]. While overall structure comparison provides some insights, the variation of just a few residues can alter activity or binding specificity, limiting functional resolution [62].
The emerging solution lies in integrating evolutionary information with structural analysis to create precise structural motifs that probe local geometric and evolutionary similarities. This approach addresses a critical need in enzymology: validating that computationally designed or annotated active sites function not just catalytically but with intended specificity. For researchers in drug development, where off-target effects can derail therapeutic candidates, this validation is paramount.
Comparative Performance Analysis of Annotation Methods
We objectively compared three methodological approaches for functional annotation using standardized benchmark datasets. The evaluation followed essential benchmarking guidelines, including clear purpose definition, comprehensive method selection, diverse datasets, and appropriate evaluation metrics [63].
Table 1: Accuracy Comparison of Functional Annotation Methods
| Method | Overall Accuracy | Accuracy at >30% Sequence Identity | Accuracy at <30% Sequence Identity | Substrate-Level Prediction Accuracy |
|---|---|---|---|---|
| ETA (Evolutionary Tracing Annotation) | 92% [62] | 99% [62] | 99% [62] | 99% (when confidence score >1) [62] |
| COFACTOR | 96% [62] | Information missing | Information missing | Information missing |
| Overall Structural Match | Information missing | Information missing | Becomes increasingly inaccurate below 45% sequence identity [62] | Information missing |
Table 2: Template Composition Analysis: ETA vs. Catalytic Residue Databases
| Residue Type | Frequency in ETA Templates | Frequency in MACiE Catalytic Database | Functional Role |
|---|---|---|---|
| Histidine | Preponderant [62] | Preponderant [62] | Catalytic |
| Aspartic Acid | Preponderant [62] | Preponderant [62] | Catalytic |
| Arginine | Preponderant [62] | Preponderant [62] | Catalytic |
| Glycine | 69% of templates [62] | Mostly absent [62] | Structural stability/dynamics |
| Proline | 27% of templates [62] | Mostly absent [62] | Structural stability/dynamics |
The data reveal that ETA maintains 99% accuracy even when sequence identity falls below 30%, outperforming both template-based matching methods like COFACTOR and overall structural matching approaches [62]. Strikingly, ETA's structural invariance remains high across varying sequence identities, with low all-atom root-mean-square deviation between templates and their cognate match sites [62].
ETA Workflow: From Query to Validation
Experimental Validation: Case Study in Carboxylesterase Prediction
Experimental Protocol
Objective: Validate ETA prediction that an uncharacterized Silicibacter sp. protein functioned as a carboxylesterase for short fatty acyl chains, despite sharing less than 20% sequence identity with known hormone-sensitive-lipase-like proteins [62].
Methodology:
Template Construction:
- Identified evolutionarily important residues through Evolutionary Tracing (ET)
- Selected 5-6 top-ranked ET residues clustering on protein surface
- Constructed 3D template representing local structural motif
Functional Assays:
- Expressed and purified wild-type Silicibacter sp. protein
- Performed enzyme activity assays with various fatty acyl chain substrates
- Measured kinetic parameters (Km, kcat) for substrate specificity profiling
- Compared activity profile with predicted hormone-sensitive-lipase-like proteins
Mutagenesis Studies:
- Designed point mutations targeting predicted motif residues
- Expressed and purified mutant protein variants
- Performed identical functional assays with mutant proteins
- Determined essential residues for catalysis and substrate specificity
Controls:
- Included positive controls (known carboxylesterases)
- Included negative controls (unrelated enzymes)
- Performed assays in triplicate with statistical analysis
Key Findings
The experimental validation confirmed that the ETA-predicted carboxylesterase activity was correct. Assays and directed mutations demonstrated that the structural motif was essential for both catalysis and substrate specificity [62]. This case study exemplifies how structural motifs of evolutionarily important residues can accurately pinpoint enzyme function even at extremely low sequence identities.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Research Reagent Solutions for Structural Validation Studies
| Reagent/Resource | Function | Specific Application |
|---|---|---|
| Evolutionary Tracing (ET) Algorithm | Ranks sequence positions by evolutionary importance | Identifies functionally critical residues for template construction [62] |
| Structural Motif Templates | 3-6 residue templates representing local structural features | Probes geometric and evolutionary similarities in protein structures [62] |
| Site-Directed Mutagenesis Kit | Creates specific point mutations in protein sequences | Tests functional importance of predicted essential residues [62] |
| Enzyme Activity Assays | Measures catalytic activity and kinetic parameters | Validates predicted enzyme function and substrate specificity [62] |
| Structural Genomics Datasets | Repository of protein structures with low sequence identity | Provides test cases for functional annotation methods [62] |
| MACiE Database | Comprehensive database of catalytic residues | Reference for comparing template composition [62] |
Integrated Workflow for Active Site Validation
Integrated Active Site Validation Workflow
Future Directions in Enzyme Validation
The field of enzyme function validation is rapidly evolving toward integrating diverse methodological approaches. Key future directions include:
Advanced Conformational Analysis: Investigating how dynamic structural elements cooperate to maintain efficient catalytic function, as demonstrated in studies of KatG catalase activity where an "Arg Switch" and oxidizable "proximal Trp" regulate function [64].
High-Throughput Computational Screening: Leveraging tools to screen hundreds of thousands of enzyme candidates for specific functions, as exemplified by ketoreductase studies that combined computational analysis with site-directed mutagenesis to understand substrate specificity [64].
Multi-functional Enzyme Characterization: Developing methods to analyze sophisticated molecular machinery in bifunctional enzymes like glutamine-hydrolyzing synthetases, which coordinate multiple catalytic activities within single protein structures [64].
The integration of computational and experimental approaches is becoming increasingly powerful, enabling both prediction and validation of structure-function relationships. Technological advances in structural techniques, spectroscopic methods, and computational tools continue to provide unprecedented insights into enzyme mechanism and function [64].
This comparison demonstrates that integrated structural analysis methods like ETA significantly outperform traditional sequence-based and overall structure-based approaches for predicting enzyme function and substrate specificity, particularly at low sequence identities. The experimental validation case study confirms that structural motifs comprising both catalytic and noncatalytic evolutionarily important residues can accurately identify enzyme activity when standard annotation methods fail.
For researchers in drug development and enzyme engineering, these approaches provide a robust framework for moving beyond catalytic residue identification toward comprehensive active site validation. By adopting these integrated methodologies, scientists can accelerate functional annotation of novel proteins while reducing errors in substrate specificity prediction—a critical advancement for applications ranging from therapeutic design to industrial biocatalysis.
Conclusion
The validation of computationally annotated enzyme function is not a single step but an integrated, iterative process that critically combines advanced AI tools with rigorous experimental confirmation. The key takeaway is that while computational methods—from generative models like ESM-MSA and ProteinGAN to specificity predictors like EZSpecificity—have dramatically improved, their predictions are not infallible and must be filtered through frameworks like COMPSS and validated with robust high-throughput assays. The future of reliable enzyme annotation and design lies in closing the loop between computation and experiment, using each round of experimental data to refine and retrain predictive models. For biomedical research, this rigorous approach is paramount; it accelerates the accurate identification of drug targets, the development of enzymatic therapeutics, and the building of trustworthy metabolic models, ultimately ensuring that foundational data driving discovery is built on a solid foundation of validated function.
References
- 1. Experimental and computational investigation of enzyme ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1009446]
- 2. Improving automatic GO annotation with semantic similarity [https://pmc.ncbi.nlm.nih.gov/articles/PMC9743508/]
- 3. Enzyme function less conserved than anticipated - PubMed [https://pubmed.ncbi.nlm.nih.gov/12051862/]
- 4. Revisiting the functional annotation of TriTryp using ... [https://www.sciencedirect.com/science/article/pii/S2405844024152747]
- 5. Computational scoring and experimental evaluation of ... [https://www.nature.com/articles/s41587-024-02214-2]
- 6. The promises and pitfalls of automated variant interpretation [https://pmc.ncbi.nlm.nih.gov/articles/PMC12513165/]
- 7. Specialty Enzymes Market Size to Reach USD 6.73 Billion ... [https://www.towardsfnb.com/insights/specialty-enzymes-market]
- 8. Prediction of enzyme function using an interpretable optimized ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc04513d]
- 9. Experimental and computational investigation of enzyme ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8491902/]
- 10. Limitations of current machine learning models in predicting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12506659/]
- 11. Improved enzyme functional annotation prediction using ... [https://www.nature.com/articles/s42003-024-07359-z]
- 12. Enzyme specificity prediction using cross-attention graph ... [https://www.nature.com/articles/s41586-025-09697-2]
- 13. Enzyme functional classification using artificial intelligence [https://www.sciencedirect.com/science/article/abs/pii/S0167779925000885]
- 14. The Missing Enzymes: A Call to Update Pharmacological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12035801/]
- 15. SARST2 high-throughput and resource-efficient protein ... [https://www.nature.com/articles/s41467-025-63757-9]
- 16. Next-Generation Protein Sequencing: Nanopore and AI ... [https://premierscience.com/pjs-25-1205/]
- 17. empowering graph neural networks with cross attention [https://www.nature.com/articles/s41598-025-88993-3]
- 18. Cross-attention graph neural networks for inferring gene ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06186-1]
- 19. Graph neural networks with configuration cross-attention ... [https://www.frontiersin.org/journals/artificial-intelligence/articles/10.3389/frai.2025.1605539/full]
- 20. Performance assessment of various graph neural network ... [https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp01072a]
- 21. Comparative accuracy of biomarkers for the prediction of ... [https://ccforum.biomedcentral.com/articles/10.1186/s13054-022-04223-6]
- 22. Discovery, design, and engineering of enzymes based on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12042125/]
- 23. A generalized platform for artificial intelligence-powered ... [https://www.nature.com/articles/s41467-025-61209-y]
- 24. Models and Methodology | BIT-LLM - iGEM 2025 [https://2025.igem.wiki/bit-llm/model]
- 25. Complete computational design of high-efficiency Kemp ... [https://rosettacommons.org/2025/08/29/complete-computational-design-of-high-efficiency-kemp-elimination-enzymes/]
- 26. Comparative Assessment of Protein Large Language Models ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06081-9]
- 27. From Machine Learning to Multimodal Models: The AI ... [https://www.sciencedirect.com/science/article/pii/S2693125725000457]
- 28. JUMPsem:从翻译后修饰数据推断酶活性的创新利器 [https://www.ebiotrade.com/newsf/2025-1/20250123165706531.htm]
- 29. Proteomics Computational Tools - NCI [https://dctd.cancer.gov/data-tools-biospecimens/calculators-computational-tools/proteomics-computational-tools]
- 30. IsobarPTM: A software tool for the quantitative analysis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3759844/]
- 31. Bioinformatics Tools and Databases for Post-Translational ... [https://www.creative-proteomics.com/proteomics/protein-science/bioinformatics-tools-databases-ptms-analysis.html]
- 32. Philosopher: a versatile toolkit for shotgun proteomics data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7509848/]
- 33. Alignment Modulates Ancestral Sequence Reconstruction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5995191/]
- 34. An experimental phylogeny to benchmark ancestral ... [https://www.nature.com/articles/ncomms12847]
- 35. Generating new protein sequences by using dense ... [https://www.aimspress.com/article/doi/10.3934/mbe.2023195?viewType=HTML]
- 36. Assessing the role of evolutionary information for ... [https://www.nature.com/articles/s41598-024-71783-8]
- 37. Improving prediction performance of general protein ... [https://www.nature.com/articles/s41467-024-52293-7]
- 38. ProteinGym: Large-Scale Benchmarks for Protein Design ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10723403/]
- 39. Experimental and computational investigation of enzyme ... [https://pubmed.ncbi.nlm.nih.gov/34555022/]
- 40. Robust Analysis of High Throughput Screening (HTS) Assay ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3727440/]
- 41. Biochemical Assay Development: Strategies to Speed Up ... [https://bellbrooklabs.com/accelerate-biochemical-assay-development/?srsltid=AfmBOoqvzk80WIjmBdXRiApl0wH6o4kZCIBfAEqyCorPP-6Yge1Aku_C]
- 42. Strategies to Optimize Protein Expression in E. coli - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7162232/]
- 43. Avoid Common Obstacles in Protein Expression [https://www.neb.com/en-us/tools-and-resources/feature-articles/bypassing-common-obstacles-in-protein-expression?srsltid=AfmBOoqNBascA2WKqOYiWQgJlQZWxS8pb2DXgCQL-aDJfhijZ_vEOVjO]
- 44. Large-scale experimental studies show unexpected amino ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3372292/]
- 45. Troubleshooting: Protein Expression [https://www.goldbio.com/blogs/articles/troubleshooting-protein-expression?srsltid=AfmBOoprekKs2HmSXc6siFYnHoKDhe7flsuUghq66Q5MqLHp9BRfGsvK]
- 46. Best Practices: Small-scale Expression Optimization [https://www.proteos.com/small-scale-expression-optimization]
- 47. Signal Peptides: From Molecular Mechanisms to Applications ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12190922/]
- 48. Disorder prediction-based construct optimization improves ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4835747/]
- 49. A half-site multimeric enzyme achieves its cooperativity ... [https://www.nature.com/articles/s41598-017-16421-2]
- 50. Monitoring Enzyme Catalysis in the Multimeric State [https://pmc.ncbi.nlm.nih.gov/articles/PMC2743789/]
- 51. A comparative analysis of 2D and 3D experimental data for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10517149/]
- 52. New AI tool helps match enzymes to substrates [https://las.illinois.edu/news/2025-11-03/new-ai-tool-helps-match-enzymes-substrates]
- 53. New AI tool helps match enzymes to substrates [https://www.igb.illinois.edu/article/new-ai-tool-helps-match-enzymes-substrates]
- 54. AI Tool EZSpecificity Accurately Predicts Enzyme-Substrate ... [https://hyper.ai/en/headlines/88006437db993c266c9c5c19fe1490a2]
- 55. Revolutionary AI Tool Streamlines Enzyme-Substrate ... [https://bioengineer.org/revolutionary-ai-tool-streamlines-enzyme-substrate-matching/]
- 56. Benchmarking Top Open-Source Speech Recognition ... [https://www.shunyalabs.ai/blog/benchmarking-top-open-source-speech-recognition-models]
- 57. Best Speech to Text Models 2025: Real-Time Agent Comparison [https://nextlevel.ai/best-speech-to-text-models/]
- 58. Innovative breakthroughs in novel image synthesis techniques ... [https://www.spiedigitallibrary.org/conference-proceedings-of-spie/13653/1365314/Innovative-breakthroughs-in-novel-image-synthesis-techniques-based-on-generative/10.1117/12.3071188.full?webSyncID=dced0db5-1d96-aaae-3719-8aac3c88c08e&sessionGUID=1c64c970-26d8-73b1-39ac-658444437c22]
- 59. Predicting Empirical AI Research Outcomes with Language ... [https://arxiv.org/html/2506.00794v1]
- 60. ICCV 2025 Open Access Repository [https://openaccess.thecvf.com/content/ICCV2025/html/Wang_IDEATOR_Jailbreaking_and_Benchmarking_Large_Vision-Language_Models_Using_Themselves_ICCV_2025_paper.html]
- 61. Speech Data Strategies for Robust ASR Performance in 2025 [https://www.futurebeeai.com/blog/speech-recognition-data-strategies-for-robust-asr]
- 62. Prediction and experimental validation of enzyme substrate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3831482/]
- 63. Essential guidelines for computational method benchmarking [https://pmc.ncbi.nlm.nih.gov/articles/PMC6584985/]
- 64. Editorial: Structure-function relationship of enzymes ... [https://www.frontiersin.org/journals/chemical-biology/articles/10.3389/fchbi.2025.1565931/full]