Automating Enzyme Discovery: A Comprehensive Snakemake Workflow for Targeted Metagenomic Screening
This article provides a complete guide for researchers and drug discovery professionals on implementing a reproducible Snakemake workflow for targeted enzyme discovery.
Automating Enzyme Discovery: A Comprehensive Snakemake Workflow for Targeted Metagenomic Screening
Abstract
This article provides a complete guide for researchers and drug discovery professionals on implementing a reproducible Snakemake workflow for targeted enzyme discovery. We begin by establishing the core bioinformatics principles, exploring key databases, and defining common targets like lipases, proteases, and polymer-degrading enzymes. The methodological section details the step-by-step construction of the pipeline, from raw metagenomic read processing and quality control to gene prediction, functional annotation, and candidate ranking. We then address common computational challenges, optimization strategies for performance and cost, and best practices for reproducibility. Finally, we cover methods for validating computational predictions through sequence analysis, structural modeling, and comparative benchmarking against existing tools. This guide empowers scientists to build robust, scalable, and transparent workflows to accelerate the identification of novel biocatalysts for industrial and therapeutic applications.
The Bioinformatics Bedrock: Core Concepts and Targets for Enzyme Discovery
Within a Snakemake-driven pipeline for targeted enzyme discovery, precise initial target definition is critical. This involves selecting enzyme classes based on desired catalytic function, relevance to a biological pathway, or industrial process suitability. This document outlines key enzyme classes, their applications, and core experimental protocols for initial characterization, forming the essential first module of a reproducible bioinformatics-to-bench workflow.
Table 1: High-Value Enzyme Classes for Targeted Discovery
| Enzyme Class (EC) | Primary Function | Key Biomedical Application | Key Industrial Application | Representative Market/Research Metric |
|---|---|---|---|---|
| Kinases (EC 2.7.11.-) | Transfer phosphate group (ATP → substrate) | Oncology (e.g., EGFR, BCR-ABL inhibitors), Inflammation | Rare; specialized biocatalysis | >50 FDA-approved drugs; ~538 human kinases mapped |
| Proteases (EC 3.4.-.-) | Hydrolyze peptide bonds | Antiviral (e.g., HIV-1 protease inhibitors), Hypertension (ACE inhibitors) | Detergent formulation, food processing (tenderization) | Global protease market ~$2.3B (2023); >60 human proteases as drug targets |
| Polymerases (EC 2.7.7.-) | Synthesize nucleic acid chains | Antiviral therapy (e.g., HCV NS5B inhibitors), Diagnostics (PCR) | Next-generation sequencing, Synthetic biology | PCR enzyme market ~$4.1B (2024); Fidelity rates: 10^-4 to 10^-6 errors/base |
| Oxidoreductases (EC 1.-.-.-) | Catalyze oxidation/reduction reactions | Antibacterial (targeting metabolic pathways), Antioxidant therapies | Bulk chemical synthesis, biosensors, biofuel cells | Global market ~$7.5B (2024); Industrial turnover numbers up to 10^6 min^-1 |
| Glycosyltransferases (EC 2.4.-.-) | Transfer sugar moieties to substrates | Antibiotic development (targeting cell wall synthesis), Glyco-engineering | Synthesis of oligosaccharides, glycoconjugates | ~200 human GT targets in disease; key for antibody-drug conjugate (ADC) development |
| Hydrolases (e.g., Lipases, EC 3.1.1.3) | Hydrolytic cleavage of esters, amides, glycosides | Digestive disorders, Lipid metabolism diseases | Bio-diesel production, enantioselective resolution, pulp/paper | Lipase market ~$735M (2024); Industrial process yields >99% enantiomeric excess |
Application Notes & Core Protocols
Application Note 1: Kinase Target Validation in a Cell Signaling Pathway Kinases are pivotal in signal transduction. Targeting specific nodes can modulate disease pathways (e.g., MAPK/ERK in cancer). A Snakemake workflow can automate the analysis of phosphoproteomics data to identify active kinase nodes from mass spectrometry output.
Protocol 1.1: In Vitro Kinase Activity Assay (Adaptable for High-Throughput Screening) Objective: Measure the phosphotransferase activity of a purified recombinant kinase candidate. Reagents: Purified kinase, specific peptide/protein substrate, ATP, Reaction Buffer (50 mM HEPES pH 7.5, 10 mM MgCl₂, 1 mM DTT), [γ-³²P]ATP or ADP-Glo Kit. Procedure:
- In a 25 µL reaction, combine 50 ng kinase, 10 µM substrate, and 10 µM ATP (with trace [γ-³²P]ATP for radiometric assay) in buffer.
- Incubate at 30°C for 30 minutes.
- Terminate Reaction:
- Radiometric: Spot reaction onto phosphocellulose paper, wash with 0.75% phosphoric acid, measure scintillation.
- Luminescent (ADP-Glo): Add ADP-Glo Reagent to deplete ATP, then add Kinase Detection Reagent to convert ADP to ATP, followed by luciferase/luciferin reaction.
- Quantify phosphate incorporation via scintillation counting or luminescence (relative light units). Data Integration: Snakemake rule can process raw luminescence/scintillation counts from plate readers into normalized activity plots.
Diagram Title: Kinase Activity Assay Workflow
Application Note 2: Hydrolase Characterization for Biocatalysis Industrial hydrolases (lipases, cellulases) require characterization of activity under process-like conditions (e.g., organic solvents, high temperature). Snakemake can manage parallel enzyme variant screening against multiple substrate and condition sets.
Protocol 2.1: pH & Temperature Profiling of Esterase/Lipase Activity Objective: Determine optimal pH and temperature for a novel hydrolase. Reagents: Purified hydrolase, p-nitrophenyl ester substrate (e.g., pNP-acetate, pNP-palmitate), Buffers (pH 4-10), Thermostatically controlled spectrophotometer. Procedure:
- Prepare 1 mM substrate in appropriate buffer (varying pH) with 1% (v/v) acetonitrile for solubility.
- Aliquot 190 µL substrate solution into a 96-well plate, pre-equilibrate at target temperature (e.g., 20°C to 70°C).
- Initiate reaction by adding 10 µL of diluted enzyme.
- Continuously monitor absorbance at 405 nm (release of p-nitrophenol) for 5 minutes.
- Calculate initial velocity (V₀) using the molar extinction coefficient of p-nitrophenol (ε₄₀₅ ≈ 16,200 M⁻¹cm⁻¹ for basic pH).
- Plot V₀ vs. pH and vs. temperature to determine optima and stability range. Workflow Integration: Snakemake rules can collate A405 time-course data from multiple plates, apply the extinction coefficient calculation, and generate comparative profile plots for hundreds of variants.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Enzyme Target Discovery & Characterization
| Reagent / Material | Primary Function in Target Research | Example Product/Kit |
|---|---|---|
| Recombinant Enzyme (Purified) | Core substrate for in vitro kinetic assays, structural studies. | His-tagged enzymes from expression systems (E. coli, insect cells). |
| Activity Assay Kits (Luminescent) | Enable high-throughput, homogeneous, non-radiometric activity measurement. | ADP-Glo Kinase Assay, Protease-Glo, CellTiter-Glo (viability). |
| Fluorogenic/Chromogenic Substrates | Provide sensitive, continuous readout of hydrolase (protease, lipase) activity. | p-Nitrophenyl (pNP) esters, AMC/Rho110-coupled peptides. |
| Phospho-Specific Antibodies | Detect and quantify phosphorylation state of kinase substrates in cell lysates. | Anti-phospho-Tyr/Ser/Thr antibodies for Western blot/ELISA. |
| Inhibitor Libraries (Small Molecule) | For target validation, selectivity screening, and drug discovery. | Published kinase inhibitor sets, protease inhibitor cocktails. |
| Thermostability Agents | Enhance enzyme stability for storage and industrial application screening. | Glycerol, trehalose, polyethylene glycols (PEGs). |
| Snakemake Workflow Manager | Automates and reproduces data analysis pipelines from raw data to figures. | Snakemake (v7.32+), conda for environment management. |
Diagram Title: Target Enzyme Discovery Thesis Workflow
Application Notes
This guide details the integration of four essential bioinformatics databases into a Snakemake workflow for targeted enzyme discovery. The workflow standardizes data retrieval and processing, enabling reproducible in silico characterization of enzyme families for applications in biotechnology and drug development.
Table 1: Core Database Comparison for Enzyme Discovery
| Database | Primary Focus | Key Data Types (Quantitative) | Relevance to Enzyme Discovery |
|---|---|---|---|
| CAZy | Carbohydrate-Active Enzymes | ~400 enzyme families; ~3M classified proteins (2024) | Identifies glycosyl hydrolases, transferases, etc., for polysaccharide degradation/synthesis. |
| MEROPS | Proteolytic Enzymes | ~4,800 peptidases; ~82,000 inhibitors (Release 12.3) | Targets protease and inhibitor discovery for therapeutic intervention. |
| BRENDA | Comprehensive Enzyme Data | ~90k enzymes; ~200k kinetic parameters (KM, kcat); ~2M EC numbers | Provides functional parameters (pH/Temp optima, kinetics) for enzyme characterization. |
| UniProt | Comprehensive Protein Data | ~220M protein entries; ~600k manually reviewed (Swiss-Prot) (2024_03) | Central source for sequence, functional annotation, and structural data cross-referencing. |
Table 2: Snakemake Workflow: Key Rule Outputs & Databases
| Rule Name | Input | Output | Primary Database Used |
|---|---|---|---|
fetch_target_families |
EC list / target | data/cazy_families.txt data/merops_clans.txt |
CAZy, MEROPS |
retrieve_sequences |
Family/Clan IDs | data/uniprot_sequences.fasta |
UniProt (via API) |
annotate_with_brenda |
Sequence IDs | data/kinetic_parameters.tsv |
BRENDA (via REST) |
generate_report |
All outputs | report/enzyme_candidates.html |
Integrated Data |
Experimental Protocols
Protocol 1: Automated Retrieval of CAZy Family Members
Objective: To programmatically obtain all protein accessions for a target Glycosyl Hydrolase (GH) family.
Materials: Snakemake workflow, Python 3.10+, biopython, requests libraries.
- Input Preparation: Create a file (
config.yaml) specifying the target CAZy family (e.g.,GH7). - Rule Execution: The Snakemake rule
fetch_cazyexecutes a Python script. - Data Fetching: Script sends an HTTP request to the CAZy API (
www.cazy.org/api). For GH7, the query is:http://www.cazy.org/api/family/GH7/proteins?format=json. - Data Parsing: Parse JSON response to extract UniProt accession numbers and protein names.
- Output: A tab-separated file (
results/cazy_GH7_members.tsv) is created, containing columns:UniProt_Acc,Protein_Name,CAZy_Family.
Protocol 2: Querying BRENDA for Kinetic Parameters
Objective: To extract kinetic data (KM, kcat) for a specified Enzyme Commission (EC) number.
Materials: Snakemake, brenda-py Python package, BRENDA license key.
- Authentication: Store BRENDA license key securely in workflow configuration.
- Rule Definition: The
query_brendarule takes an EC number (e.g.,3.2.1.176) as input. - Parameter Extraction: Using
brenda-py, query for allKM_VALUEandTURNOVER_NUMBERparameters. - Data Filtering: Filter results by organism (e.g.,
Homo sapiens) and substrate. Calculate mean ± SD for replicates. - Output: A structured file (
results/brenda_kinetics_3.2.1.176.tsv) with columns:Substrate,KM_mM,kcat_per_s,Organism,Reference.
Protocol 3: Integrated Annotation Pipeline via Snakemake
Objective: To unify data from CAZy, MEROPS, and BRENDA for a target enzyme sequence via its UniProt ID. Materials: Integrated Snakemake workflow, all listed databases.
- Start Point: Input a UniProt ID (e.g.,
P00784) into the workflow. - Parallel Queries: The workflow triggers concurrent rules:
- Rule A: Maps ID to CAZy family via CAZy-UniProt cross-reference.
- Rule B: Maps ID to MEROPS peptidase family via MEROPS flat-file download.
- Rule C: Fetches all kinetic data for the protein's EC number from BRENDA.
- Data Aggregation: A consolidation rule (
aggregate_annotations) merges all results into a single JSON document. - Validation: Check for conflicting annotations (e.g., CAZy family vs. MEROPS clan).
- Final Output: A comprehensive annotation file (
results/{uniprot_id}_full_annotation.json) ready for candidate prioritization.
Visualizations
Database Integration in Snakemake Workflow
Protocol: Kinetic Data from BRENDA
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Digital Tools & Resources for the Workflow
| Item / Resource | Function / Description | Source / Example |
|---|---|---|
| Snakemake | Workflow management system to create reproducible and scalable data analyses. | https://snakemake.github.io |
| Biopython | Python library for biological computation; used for parsing FASTA, GenBank, and API calls. | https://biopython.org |
| Brenda-py | Official Python client for programmatically querying the BRENDA database. | https://pypi.org/project/brenda-py |
| CAZy API | Programmatic interface (REST) to retrieve CAZy family and protein data in JSON format. | http://www.cazy.org/api |
| MEROPS Flatfiles | Regularly updated text files containing all peptidase and inhibitor data for local parsing. | FTP: ftp.ebi.ac.uk/pub/databases/merops |
| UniProt REST API | Web service to retrieve protein data in various formats (JSON, XML, FASTA) by accession. | https://www.uniprot.org/help/api |
| Conda/Mamba | Package and environment management to ensure dependency stability across analyses. | https://mamba.readthedocs.io |
| Graphviz (Dot) | Open-source graph visualization software used to render workflow diagrams. | https://graphviz.org |
Application Notes: Snakemake in Targeted Enzyme Discovery
Within a thesis focused on developing a Snakemake workflow for targeted enzyme discovery, the platform's core features directly address critical research bottlenecks. Enzyme discovery pipelines involve sequential, data-dependent steps—genome mining, sequence analysis, homology modeling, cloning, expression, and functional assays. Snakemake orchestrates these steps into a single, reproducible, and scalable computational workflow.
Table 1: Quantitative Impact of Adopting Snakemake for Bioinformatics Workflows
| Metric | Traditional Scripting | Snakemake Workflow | Improvement |
|---|---|---|---|
| Reproducibility | Manual documentation; hard-coded paths | Declarative, version-controlled rule definitions | High (Automated environment & data provenance) |
| Pipeline Execution Time | Linear, manual step triggering | Automatic parallelization of independent jobs | Up to 90% reduction on multi-core systems |
| Computational Resource Use | Often underutilized | Efficient, configurable resource management (cores, memory) | 30-70% more efficient |
| Error Recovery | Full restart or manual intervention | Automatic checkpointing and partial re-runs | Saves >50% time after mid-pipeline failures |
| Scalability (HPC/Cloud) | Requires extensive manual modification | Native support for cluster and cloud execution | Seamless transition from laptop to cluster |
Detailed Experimental Protocols
Protocol 1: Snakemake Workflow for In Silico Enzyme Candidate Identification
This protocol details the computational steps for identifying putative hydrolase enzymes from metagenomic data, a common first step in discovery pipelines.
- Input Preparation: Place raw paired-end metagenomic reads (e.g.,
sample_R1.fastq,sample_R2.fastq) in the/data/rawdirectory. Create a sample sheet (samples.tsv) mapping sample IDs to file paths. - Quality Control & Assembly:
- Implement a Snakemake rule using
FastQC(v0.12.1) for initial quality reports. - Implement a rule using
Trimmomatic(v0.39) to remove adapters and low-quality bases (parameters: ILLUMINACLIP:TruSeq3-PE.fa:2:30:10, LEADING:3, TRAILING:3, SLIDINGWINDOW:4:15, MINLEN:36). - Implement a rule for de novo assembly using
MEGAHIT(v1.2.9) with default parameters.
- Implement a Snakemake rule using
- Open Reading Frame (ORF) Prediction: Implement a rule using
Prodigal(v2.6.3) in meta-mode (-p meta) on the assembled contigs (*.fa). Output will be nucleotide (*.fna) and amino acid (*.faa) sequence files. - Homology-Based Screening: Implement a rule using
HMMER(v3.3.2) to search the*.faaproteins against thePfamdatabase. Use a curated Hidden Markov Model (HMM) profile for the enzyme family of interest (e.g.,PF07859for alpha/beta hydrolase fold). Retain hits with an e-value< 1e-10. - Output Consolidation: A final rule collates all high-confidence hits into a single, annotated FASTA file (
final_candidates.faa) with associated metadata (sample origin, contig length, HMM score).
Protocol 2: Snakemake-Driven Structural Characterization Pipeline
This protocol follows Protocol 1, taking candidate sequences for 3D structure prediction and analysis.
- Input: The
final_candidates.faafile from Protocol 1. - Multiple Sequence Alignment (MSA): Implement a rule using
Clustal Omega(v1.2.4) orMAFFT(v7.505) to generate an MSA of candidate sequences against known reference structures. - Homology Modeling: Implement a rule using
MODELLER(v10.4) or a wrapper forSWISS-MODEL. The rule downloads a suitable template (e.g., PDB ID: 1EXA) and generates 5 models per candidate. - Model Evaluation: Implement a rule using
DOPEorQMEANscores within MODELLER to select the best model for each candidate. - Active Site Analysis: Implement a rule using
PyMOLorfpocketto analyze the predicted model's active site cavity, generating report files.
Mandatory Visualization
Diagram 1: Snakemake Enzyme Discovery Workflow
Diagram 2: Snakemake Rule Dependency Logic
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools & Resources for Snakemake-Driven Enzyme Discovery
| Item | Function in Workflow | Example/Note |
|---|---|---|
| Snakemake (v7.32+) | Core workflow management system. Enforces reproducible and scalable pipeline execution. | Must be installed via Conda/Mamba. |
| Conda/Mamba | Environment and package manager. Creates isolated software environments for each pipeline step. | Critical for reproducibility of tool versions. |
| HMMER Suite | Scans protein sequences against profile HMM databases (e.g., Pfam) to identify enzyme families. | hmmsearch is the key command. |
| Prodigal | Predicts protein-coding genes (ORFs) in microbial (meta)genomic sequences. | Operates in -p meta mode for complex communities. |
| MEGAHIT | Efficient de novo assembler for large and complex metagenomic datasets. | Used for uncultivable microbial samples. |
| MODELLER | Generates 3D homology models of protein structures based on known templates. | Requires a PDB template of a related enzyme. |
| Conda Forge/Bioconda | Community repositories providing >10,000 bioinformatics packages as Conda recipes. | Primary source for installing tools within Snakemake. |
| Pfam Database | Curated collection of protein family HMM profiles. Used for functional annotation. | Profile for target enzyme family (e.g., hydrolase) is required. |
| Git | Version control for the Snakemake workflow script (Snakefile), configs, and analysis code. |
Tracks all changes to the computational methods. |
| Singularity/Apptainer | Containerization platform. Used with Snakemake to encapsulate entire software environments. | Ensures perfect reproducibility on HPC/clusters. |
Metagenomics has revolutionized our ability to access the genetic potential of uncultured microorganisms from any environment. This approach functions as a powerful discovery engine, bypassing the need for cultivation by extracting and sequencing DNA directly from complex samples like soil, ocean water, or the human gut. The resulting sequence data is assembled and binned to reconstruct genomes or gene fragments, culminating in extensive gene catalogs. These catalogs are treasure troves for identifying novel enzymes, biosynthetic pathways, and bioactive compounds with applications in drug discovery, industrial biocatalysis, and bioremediation.
Within the context of a targeted enzyme discovery thesis, a reproducible and scalable bioinformatics workflow is paramount. The Snakemake workflow management system provides an ideal framework for constructing a pipeline that standardizes the process from raw sequencing reads to a curated gene catalog, enabling the identification of target enzyme families (e.g., glycoside hydrolases, polyketide synthases) across multiple metagenomic datasets.
Key Protocols for Metagenomic Gene Catalog Construction
Protocol 2.1: Environmental DNA Extraction and Library Preparation
Objective: To obtain high-quality, high-molecular-weight DNA from a complex environmental sample suitable for shotgun metagenomic sequencing. Materials: See The Scientist's Toolkit (Section 5). Method:
- Sample Collection & Stabilization: Collect sample (e.g., 1g soil, 200ml water filtered onto 0.22μm membrane). Immediately freeze in liquid nitrogen or preserve in a DNA/RNA stabilization buffer.
- Cell Lysis: Use a combination of mechanical (bead-beating), chemical (lysozyme, SDS), and thermal lysis. For soil, subject sample to bead-beating in lysis buffer for 45 seconds at 6.0 m/s.
- DNA Purification: Remove contaminants (humic acids, proteins) using CTAB or dedicated commercial kit columns. Perform precipitations with isopropanol.
- Quality Assessment: Verify DNA integrity via pulsed-field gel electrophoresis (>20 kb ideal) and quantify using Qubit fluorometry. Ensure A260/A280 ratio is ~1.8.
- Library Preparation: Fragment DNA to ~350 bp (e.g., using ultrasonication). Perform end-repair, A-tailing, and adapter ligation using a commercial kit (e.g., Illumina TruSeq). Amplify library with limited-cycle PCR.
- Sequencing: Pool libraries and sequence on an Illumina NovaSeq platform to a target depth of ≥50 million 150-bp paired-end reads per sample.
Protocol 2.2: Snakemake Workflow for Read Processing, Assembly, and Gene Prediction
Objective: To deploy a standardized, reproducible pipeline for converting raw reads into a predicted protein catalog.
Prerequisites: Install Snakemake, conda, and required bioinformatics tools (FastQC, Trimmomatic, MEGAHIT, metaSPAdes, MetaGeneMark, Prokka).
Workflow Configuration (config.yaml):
Core Snakemake Rulefile (Snakefile):
Execution: Run the workflow with snakemake --cores 16 --use-conda.
Protocol 2.3: Targeted Enzyme Discovery from Gene Catalogs
Objective: To screen the assembled gene catalog for sequences homologous to a target enzyme family. Method:
- Catalog Compilation: Concatenate all per-sample
.faafiles into a non-redundant catalog usingcd-hitat 95% identity (cd-hit -i catalog.faa -o catalog_nr.faa -c 0.95). - Homology Search: Create a Hidden Markov Model (HMM) profile from a multiple sequence alignment of known target enzymes (e.g., from PFAM). Search the catalog against this HMM using
hmmsearch(hmmsearch --cpu 8 --tblout hits.txt pfam_profile.hmm catalog_nr.faa). - Functional Annotation: Annotate candidate hits with known domains using
hmmscanagainst the PFAM database. - Phylogenetic Analysis: Align candidate sequences with references using MAFFT, build a phylogenetic tree with FastTree, and visualize to identify novel clades.
- Priority Ranking: Rank candidates based on sequence completeness, novelty (distance to known sequences), and domain architecture complexity.
Table 1: Representative Metagenomic Sequencing and Assembly Statistics
| Metric | Soil Sample | Marine Sample | Human Gut Sample | Unit |
|---|---|---|---|---|
| Sequencing Depth | 80 | 60 | 100 | Million reads |
| Raw Data | 24 | 18 | 30 | GB |
| Post-QC Reads | 76.5 | 78.2 | 82.1 | % retained |
| Number of Contigs | 1,200,000 | 850,000 | 450,000 | Count (>500 bp) |
| N50 Contig Length | 2,450 | 3,100 | 5,800 | Base pairs |
| Predicted Genes | 1.8 | 1.2 | 0.9 | Million |
| Non-Redundant Genes | 1.1 | 0.8 | 0.6 | Million (95% ID) |
Table 2: Enzyme Discovery Yield from a 10-Sample Soil Metagenome Catalog
| Target Enzyme Class | PFAM Profile | Candidate Hits | Complete ORFs* | Novel Clades |
|---|---|---|---|---|
| Glycoside Hydrolases | PF00150 (Cel5) | 1,250 | 890 | 3 |
| Proteases (Serine) | PF00089 (Trypsin) | 680 | 550 | 2 |
| Polyketide Synthases | PF00109 (KS domain) | 95 | 42 | 5 |
| Laccases | PF00394 (Cu-oxidase) | 310 | 180 | 1 |
| Esterases/Lipases | PF00151 (Lipase) | 720 | 610 | 4 |
Open Reading Frame with start and stop codon. *Monophyletic branch containing only environmental sequences.
Mandatory Visualizations
Metagenomics Discovery Pipeline Overview
Snakemake for Reproducible Gene Catalog
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Metagenomic Workflows
| Item | Function & Rationale | Example Product |
|---|---|---|
| DNA/RNA Shield | Immediate biological sample preservation at point of collection. Inactivates nucleases and stabilizes community profile. | Zymo Research DNA/RNA Shield |
| Magnetic Bead-Based Cleanup Kits | High-throughput, automatable purification of DNA from complex samples, removing PCR inhibitors (humics, polyphenols). | MagMAX Microbiome Ultra Kit |
| Ultra-high-quality DNA Polymerase | Accurate amplification of metagenomic libraries with minimal bias, essential for representing low-abundance species. | KAPA HiFi HotStart ReadyMix |
| Unique Dual Index (UDI) Kits | Multiplexing of hundreds of samples without index crosstalk, enabling large-scale comparative studies. | Illumina IDT for Illumina UDIs |
| Long-read Sequencing Chemistry | Resolving complex repeats and producing complete microbial genomes from metagenomes. | PacBio HiFi sequencing |
| HMMER Software Suite | Sensitive profile Hidden Markov Model searches for detecting distant homologs of target enzyme families in gene catalogs. | HMMER v3.4 |
| cd-hit Suite | Clustering of predicted protein sequences to generate non-redundant gene catalogs, reducing downstream analysis complexity. | cd-hit v4.8.1 |
This application note details the establishment of explicit criteria for enzyme candidate selection within a Snakemake workflow for targeted enzyme discovery. The reproducibility and scalability of the automated workflow depend on the precise, programmatic definition of success at each stage: Specificity (target engagement), Activity (catalytic function), and Expression (biochemical yield). These criteria form the decision gates within the workflow's directed acyclic graph (DAG).
Table 1: Specificity & Binding Criteria
| Criterion | Target Metric | Threshold Value | Experimental Method |
|---|---|---|---|
| Binding Affinity | Dissociation Constant (Kd) | ≤ 10 µM | Surface Plasmon Resonance (SPR) |
| Target Selectivity | Fold-Change vs. Off-Targets | ≥ 100-fold | Thermal Shift Assay (ΔTm) |
| Theoretical Specificity | Predicted Docking Score | ≤ -7.0 kcal/mol | In silico Molecular Docking |
Table 2: Enzymatic Activity Criteria
| Criterion | Target Metric | Threshold Value | Notes |
|---|---|---|---|
| Catalytic Efficiency | kcat/KM | ≥ 10^3 M^-1s^-1 | Initial screen |
| Thermostability | T_m (Melting Temp) | ≥ 55°C | For industrial relevance |
| pH Stability | % Activity Retention (pH 4-9) | ≥ 70% | Broad-range activity |
Table 3: Expression & Solubility Criteria
| Criterion | Target Metric | Threshold Value | Assessment Point |
|---|---|---|---|
| Soluble Yield | Protein Concentration | ≥ 0.5 mg/L culture | After affinity purification |
| Purity | Single Band on SDS-PAGE | ≥ 90% | Post-purification analysis |
| Aggregation State | Monomeric Peak (%) | ≥ 85% | Size-Exclusion Chromatography |
Experimental Protocols
Protocol 3.1: High-Throughput Thermal Shift Assay for Specificity
- Objective: Quantify target binding and selectivity via ligand-induced thermal stabilization.
- Reagents: Purified enzyme (2 µM), target ligand (10 µM), SYPRO Orange dye (5X), off-target ligands.
- Procedure:
- In a 96-well PCR plate, mix 18 µL of enzyme solution with 2 µL of ligand (or buffer control).
- Add 5 µL of 5X SYPRO Orange dye. Final reaction volume: 25 µL.
- Perform a temperature ramp from 25°C to 95°C at 1°C/min in a real-time PCR instrument.
- Monitor fluorescence intensity (excitation/emission ~470/570 nm).
- Calculate melting temperature (Tm) from the first derivative of the fluorescence curve.
- Determine ΔTm (Tm,with ligand - Tm,buffer). A ΔTm ≥ 2°C indicates significant binding. Selectivity is confirmed if ΔTm for target is ≥ 2°C and for off-targets is < 1°C.
Protocol 3.2: Microtiter Plate-Based Kinetic Assay (kcat/KM)
- Objective: Determine initial catalytic efficiency.
- Reagents: Purified enzyme, chromogenic/fluorogenic substrate, assay buffer.
- Procedure:
- Prepare substrate solutions across a 8-point concentration range (e.g., 0.1-5 x K_M).
- In a 96-well plate, add 80 µL of substrate solution per well. Pre-incubate at assay temperature (e.g., 30°C).
- Initiate reaction by adding 20 µL of diluted enzyme. Final volume: 100 µL.
- Immediately monitor product formation by absorbance/fluorescence every 10-30 seconds for 5-10 minutes.
- Fit initial linear rates (v0) to the Michaelis-Menten equation (v0 = (Vmax * [S]) / (KM + [S])) using nonlinear regression software (e.g., GraphPad Prism).
- Calculate kcat = Vmax / [Enzyme]. Report kcat/KM.
Mandatory Visualizations
Diagram 1: Snakemake DAG for Enzyme Discovery
Diagram 2: Specificity-Activity-Expression Funnel
The Scientist's Toolkit: Research Reagent Solutions
| Reagent/Material | Supplier Examples | Function in Discovery Workflow |
|---|---|---|
| HisTrap HP Columns | Cytiva, Qiagen | Immobilized metal affinity chromatography (IMAC) for high-yield purification of His-tagged recombinant enzymes. |
| Chromogenic Substrates (pNP-esters) | Sigma-Aldrich, Tokyo Chemical Industry | Hydrolyzable substrates that release para-nitrophenol (yellow), enabling rapid, spectrophotometric activity screens. |
| SYPRO Orange Protein Gel Stain | Thermo Fisher Scientific | Environment-sensitive dye used in Thermal Shift Assays to monitor protein unfolding and ligand binding. |
| Biacore CM5 Sensor Chips | Cytiva | Gold surface for covalent immobilization of target molecules for real-time, label-free binding kinetics via SPR. |
| HiLoad 16/600 Superdex 200 pg | Cytiva | High-resolution size-exclusion chromatography column for assessing protein purity, aggregation state, and oligomerization. |
| IPTG (Isopropyl β-d-1-thiogalactopyranoside) | GoldBio, Thermo Fisher | Chemical inducer for lac/tac promoter systems in E. coli expression, triggering recombinant protein production. |
Building the Pipeline: A Step-by-Step Snakemake Workflow from Reads to Candidates
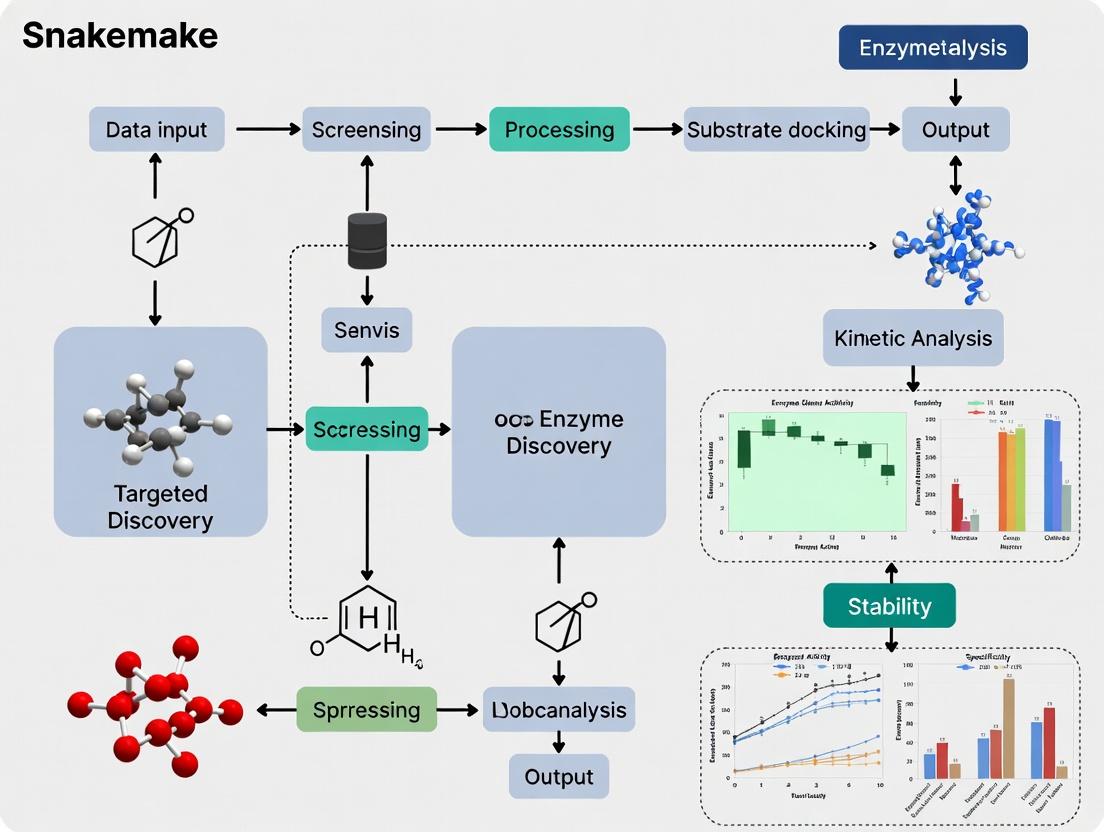
1. Introduction and Application Notes Within a thesis on developing a Snakemake workflow for targeted enzyme discovery, visualizing the computational pipeline as a Directed Acyclic Graph (DAG) is critical. The DAG provides a formal, reproducible representation of data provenance, rule dependencies, and execution order. This protocol details the creation and interpretation of such a DAG for a standard enzyme discovery pipeline, which integrates genomic data mining, homology modeling, and functional prediction.
2. Protocol: Generating and Interpreting the Snakemake DAG
- Objective: To generate a visual DAG of the enzyme discovery workflow and interpret key components.
- Software: Snakemake (v7+), Graphviz (system installation), Python.
- Procedure:
- Workflow Design: Implement a Snakemake workflow (
Snakefile) with rules for each discovery step (see Table 1). - DAG Generation: In the terminal, execute:
snakemake --dag | dot -Tpng > workflow_dag.png. - Interpretation: Analyze the generated PNG file. Rectangular nodes represent jobs (rule execution), directed edges represent dependencies, and oval nodes represent input/output files.
- Workflow Design: Implement a Snakemake workflow (
- Key Output: A DAG diagram visualizing the entire workflow's data flow and rule hierarchy.
3. Workflow DAG Visualization The core enzyme discovery pipeline DAG, generated from the Snakemake rules, is depicted below.
Diagram Title: Snakemake DAG for Enzyme Discovery Pipeline
4. Core Snakemake Rules for Enzyme Discovery Table 1: Summary of Key Workflow Rules and Data Flow
| Rule Name | Input(s) | Output(s) | Software/Tool | Core Function |
|---|---|---|---|---|
BLAST_homologs |
metagenome_db.fasta, query.faa |
homologs.fasta |
DIAMOND/BLAST | Identifies homologous sequences from metagenomic data. |
build_msa |
homologs.fasta |
alignment.clustal |
Clustal Omega/MAFFT | Generates multiple sequence alignment of homologs. |
build_hmm |
alignment.clustal |
profile.hmm |
HMMER | Creates a Hidden Markov Model profile for sensitive searches. |
comparative_modeling |
profile.hmm, templates.pdb |
models/ (PDB files) |
MODELLER, SWISS-MODEL | Builds 3D protein structure models. |
predict_active_sites |
models/ (PDB files) |
active_sites.tsv |
CASTp, DeepSite | Predicts catalytic and binding pockets from 3D models. |
generate_report |
All major outputs (homologs.fasta, active_sites.tsv, etc.) |
discovery_report.pdf |
Pandas, Matplotlib, ReportLab | Compiles results into a comprehensive summary document. |
5. Protocol: Active Site Prediction from Homology Models
- Objective: To identify and characterize putative active sites from computationally modeled enzyme structures.
- Reagents/Materials: Homology model in PDB format, computing cluster or workstation.
- Software: CASTp 3.0 (web server or local) or DeepSite (DL-based).
- Procedure (using CASTp):
- Input Preparation: Ensure your homology model PDB file is correctly formatted (ATOM records only).
- Submission: Upload the PDB file to the CASTp web server (http://sts.bioe.uic.edu/castp/).
- Parameter Setting: Set the probe radius to 1.4 Å (approximating a water molecule). Use default settings for other parameters.
- Execution: Run the analysis. The server will calculate surface-accessible and molecular-surface pockets.
- Analysis: Review the ranked list of predicted pockets. Identify the largest pocket with complementary chemical features (e.g., proximity to conserved catalytic residues from the MSA). Export the coordinates and volume data.
- Expected Output: A table of predicted binding pockets with metrics (volume, area, residues).
6. The Scientist's Toolkit: Key Research Reagent Solutions Table 2: Essential Digital and Computational Tools for the Workflow
| Item | Function/Benefit | Example/Provider |
|---|---|---|
| Snakemake | Workflow management system enabling reproducible, scalable, and modular data analysis pipelines. | https://snakemake.github.io/ |
| DIAMOND | Ultra-fast protein sequence aligner for BLAST-like searches against large metagenome databases. | https://github.com/bbuchfink/diamond |
| HMMER | Suite for profiling with Hidden Markov Models, essential for sensitive remote homology detection. | http://hmmer.org/ |
| SWISS-MODEL | Fully automated, web-based protein structure homology-modelling server. | https://swissmodel.expasy.org/ |
| CASTp | Computes topographic descriptors of protein structures, identifying and measuring binding pockets. | University of Illinois at Chicago |
| Conda/Bioconda | Package and environment manager crucial for installing and maintaining consistent bioinformatics tool versions. | https://conda.io/, https://bioconda.github.io/ |
| Jupyter Notebook | Interactive environment for exploratory data analysis, visualization, and documenting interim results. | https://jupyter.org/ |
Application Notes
Within the thesis project "A Scalable Snakemake Workflow for High-Throughput Targeted Enzyme Discovery," the initial preprocessing of raw sequencing reads is the critical first step that determines downstream analytical success. This stage ensures that low-quality data, adapter contamination, and sequencing artifacts do not bias subsequent assembly, annotation, and functional screening for novel biocatalysts. The implementation of Rule 1 as a modular, reproducible Snakemake rule guarantees consistent quality assessment and trimming across hundreds of metagenomic or transcriptomic samples typical in discovery campaigns.
Recent benchmarking studies (2023-2024) indicate that rigorous preprocessing improves functional gene annotation accuracy by 15-25% and reduces false-positive variant calls in heterogeneous environmental samples. The integrated use of FastQC for diagnostic reporting, Trimmomatic for adaptive trimming, and MultiQC for aggregated visualization is now considered the standard for robust NGS pipelines in industrial enzyme discovery.
Table 1: Impact of Preprocessing Parameters on Enzyme Discovery Metrics
| Parameter | Typical Value | Post-Trim Mean Read Length (bp) | Gene Call Recovery Rate (%) | Effect on Downstream Assembly (N50) |
|---|---|---|---|---|
| No Trimming | N/A | 150 | 100 (Baseline) | 1.2 Mb |
| SLIDINGWINDOW (4:20) | Default | 132 | 98.5 | 1.5 Mb |
| SLIDINGWINDOW (4:30) | Aggressive | 118 | 96.8 | 1.7 Mb |
| MINLEN (36) | Standard | 130 | 98.2 | 1.55 Mb |
| MINLEN (75) | Stringent | 140 | 97.1 | 1.65 Mb |
Experimental Protocols
Protocol 1.1: Initial Quality Assessment with FastQC
Objective: Generate per-base sequence quality, adapter contamination, and GC content reports for raw FASTQ files.
- Input: Paired-end or single-end raw FASTQ files (
*.fastq.gz). - Software: FastQC v0.12.1.
- Command:
- Output Interpretation: Examine
fastqc_report.html. Key warnings requiring Trimmomatic intervention include:- "Per base sequence quality" falling below Phred score 20 in later cycles.
- "Adapter Content" exceeding 5%.
- "Overrepresented sequences" > 1% of total.
Protocol 1.2: Read Trimming and Filtering with Trimmomatic
Objective: Remove adapters, low-quality bases, and discard short reads.
- Input: Raw FASTQ files.
- Software: Trimmomatic-0.39.
- Reagents/Materials:
- Adapter FASTA file (e.g.,
TruSeq3-PE-2.fafor Illumina). - Java Runtime Environment (JRE).
- Adapter FASTA file (e.g.,
- Command for Paired-End Reads:
- Parameters Justification for Enzyme Discovery:
ILLUMINACLIP:2:30:10: Removes adapters with high stringency to prevent false gene fusion.SLIDINGWINDOW:4:25: Balances quality retention with removal of error-prone regions.MINLEN:36: Ensures reads are long enough for open-reading-frame prediction.
Protocol 1.3: Aggregated Reporting with MultiQC
Objective: Compile all FastQC and Trimmomatic reports into a single interactive document.
- Input: All
fastqc_data.txt,summary.txt, and Trimmomatic log files. - Software: MultiQC v1.14.
- Command:
- Output Use: The
multiqc_report.htmlprovides a cohort-level view, enabling batch effect detection critical for comparative metagenomics in enzyme discovery.
Visualizations
Diagram Title: Snakemake Preprocessing & QC Workflow
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for NGS Preprocessing
| Item | Function in Preprocessing | Example/Supplier |
|---|---|---|
| Illumina Adapter Sequences | Specifies oligonucleotide sequences for Trimmomatic to identify and remove. Required for accurate read trimming. | File: TruSeq3-PE-2.fa (bundled with Trimmomatic). |
| High-Quality Reference Genome (Optional) | For quality control alignment to assess read integrity in known systems. | e.g., E. coli K-12 spike-in control genome. |
| Phred Score Calibration Libraries | Validates base-calling accuracy of the sequencing platform used. | PhiX Control v3 (Illumina). |
| Computational Resources | Adequate RAM and CPU for in-memory sorting of reads during trimming. | Minimum 16GB RAM, 4+ cores per sample. |
| Sample Tracking Sheet | Critical metadata linking sample ID to sequencing lane and library prep. | CSV file with headers: SampleID, Lane, AdapterSet. |
Application Notes
This section details the integration of metagenomic assembly and binning tools into a unified Snakemake workflow designed for targeted enzyme discovery. Efficient recovery of high-quality metagenome-assembled genomes (MAGs) is critical for identifying novel biosynthetic gene clusters and catalytic proteins from complex microbial communities.
Comparative Analysis of Assembly and Binning Tools: The performance of assemblers and binners varies with dataset characteristics such as read length, community complexity, and sequencing depth. The following table summarizes key quantitative metrics from recent benchmark studies, guiding tool selection within the workflow.
Table 1: Performance Metrics for Assembly and Binning Tools
| Tool Name | Primary Use | Key Metric (NGA50)* | Key Metric (Completeness)* | Key Metric (Contamination)* | Optimal Use Case |
|---|---|---|---|---|---|
| MEGAHIT | De novo assembly | High (for complex communities) | N/A | N/A | Large, complex communities; compute/memory efficient. |
| metaSPAdes | De novo assembly | Very High (for isolate-like samples) | N/A | N/A | Lower-complexity communities or high-depth datasets. |
| MaxBin 2.0 | Contig binning | N/A | 70-90% | 0-10% | Binning based on sequence composition and abundance. |
*Representative ranges from benchmark studies; actual values are dataset-dependent.
Experimental Protocols
Protocol 1: Metagenomic Co-Assembly with MEGAHIT and metaSPAdes
This protocol describes the assembly of quality-filtered, host-depleted paired-end reads into contigs.
Materials:
- Input: Interleaved or paired forward/reverse FASTQ files from multiple samples (e.g.,
sample1_qc_R1.fq.gz,sample1_qc_R2.fq.gz). - Hardware: Multi-core server with ≥64 GB RAM for moderate-sized datasets.
- Software: MEGAHIT (v1.2.9) or metaSPAdes (v3.15.5) installed via Conda.
Method:
- Tool Selection: For a typical multi-sample enzyme discovery project, MEGAHIT is recommended for its efficiency. metaSPAdes can be used for higher-quality, less complex samples.
- Co-assembly Execution: For MEGAHIT: For metaSPAdes:
- Output: The primary contig file is
./assembly_output/final_contigs.fasta(MEGAHIT) or./assembly_output/contigs.fasta(metaSPAdes).
Protocol 2: Contig Binning with MaxBin 2.0
This protocol recovers draft genomes (MAGs) from assembled contigs using sequence composition and sample-specific abundance profiles.
Materials:
- Input: Assembled contigs (
final_contigs.fasta). - Input: Read abundance files in
.abundformat for each sample, generated by aligning reads back to contigs (e.g., using Bowtie2 andcoverm). - Software: MaxBin 2.0 (v2.2.7), Bowtie2,
coverm.
Method:
- Generate Abundance Profiles:
- Execute MaxBin Binning:
Where
abundance_filelist.txtcontains paths to all.abundfiles. - Output: Binned contigs in FASTA format (e.g.,
maxbin_output.001.fasta). Each file represents one draft MAG.
Protocol 3: Integrated Snakemake Rule Definition
This protocol integrates the above steps into a reproducible Snakemake rule for the thesis workflow.
Materials: Snakemake (v7+), Conda environment with required tools.
Method:
- Define the rule in the
Snakefile:
Mandatory Visualization
Title: Metagenomic Assembly and Binning Workflow for Enzyme Discovery
The Scientist's Toolkit
Table 2: Essential Research Reagents and Materials for Metagenomic Assembly & Binning
| Item | Function/Application | Notes for Workflow Integration |
|---|---|---|
| High-Quality DNA Extraction Kit (e.g., DNeasy PowerSoil Pro) | Inhibitor-free DNA isolation from environmental samples. | Critical for long, accurate reads; input for library prep. |
| Illumina-Compatible Library Prep Kit | Prepares sequencing libraries from metagenomic DNA. | Ensure insert size is optimal for assembler (e.g., 300-800bp). |
| Bowtie2 | Fast, sensitive read alignment for generating coverage profiles. | Used in Snakemake rule to map reads back to contigs for binning. |
| CheckM | Assesses completeness and contamination of recovered MAGs. | Essential QC step after binning to filter low-quality MAGs. |
Conda Environment (metagenomics.yaml) |
Manages precise software versions for reproducibility. | Includes MEGAHIT, metaSPAdes, MaxBin2, Bowtie2, coverm. |
| High-Performance Computing Cluster | Provides necessary CPU cores and RAM for assembly steps. | MEGAHIT is memory-efficient; metaSPAdes requires significant RAM. |
Application Notes
Within a Snakemake workflow for targeted enzyme discovery, accurate gene prediction and ORF calling are critical first steps in converting assembled metagenomic or genomic sequences into actionable protein candidates. This step transforms contiguous DNA sequences (contigs) into a standardized protein catalog, which serves as the input for downstream homology searches, domain analysis, and functional screening.
Prodigal is a fast, prokaryotic gene-finding tool widely used for bacterial and archaeal genomes and metagenomes. It is favored for its speed and accuracy in identifying coding sequences without prior training.
MetaGeneMark employs a hidden Markov model (HMM) trained on metagenomic sequences, making it particularly robust for diverse, fragmented, and unknown microbial communities often encountered in environmental samples.
The choice between tools depends on the sample origin. For well-characterized, single-organism genomes, Prodigal is typically sufficient. For complex metagenomes, MetaGeneMark or a consensus approach may yield a more complete gene set. Integrating both tools and comparing results can increase confidence in predictions, especially for novel enzymes where gene boundaries are ambiguous.
Protocols
Protocol 1: ORF Prediction with Prodigal in a Snakemake Context
Objective: Predict protein-coding genes from assembled contigs.
Input: {sample}.contigs.fasta
Output: {sample}_prodigal.faa (protein sequences), {sample}_prodigal.gff (gene coordinates).
Snakemake Rule Definition:
Conda Environment (
envs/prodigal.yaml):
Protocol 2: ORF Prediction with MetaGeneMark in a Snakemake Context
Objective: Predict genes using models optimized for metagenomes.
Input: {sample}.contigs.fasta
Output: {sample}_mgm.faa, {sample}_mgm.gff
Download Model File: First, download the appropriate model file (
MetaGeneMark_v1.mod) from http://topaz.gatech.edu/GeneMark/license_download.cgi (requires registration). Place it in aresources/directory.Snakemake Rule Definition:
Conda Environment (
envs/mgm.yaml):
Protocol 3: Consensus Prediction Workflow
Objective: Merge predictions from both tools to generate a high-confidence non-redundant protein set. Strategy: Use CD-HIT to cluster proteins from both tools at 100% identity, then select the longest representative sequence per cluster.
- Snakemake Rule for Clustering:
Table 1: Comparison of Gene Prediction Tools for Enzyme Discovery
| Feature | Prodigal | MetaGeneMark |
|---|---|---|
| Primary Use Case | Isolated prokaryotic genomes | Metagenomic/metatranscriptomic assemblies |
| Underlying Model | Dynamic programming (Markov models) | Hidden Markov Model (HMM) |
| Speed | Very Fast | Moderate |
| Metagenome Mode | Yes (-p meta) |
Native |
| Typical Outputs | FAA (proteins), GFF (coordinates), NT (nucleotide genes) | FAA, GFF |
| Key Advantage | Speed, low false positive rate | Accuracy on short, novel fragments |
| Recommendation in Workflow | Default for isolate data | Use for complex environmental samples |
Visualizations
Title: Consensus Gene Prediction & ORF Calling Workflow
Title: Snakemake Rule Integration for Gene Prediction
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for Gene Prediction & ORF Calling
| Item | Function in Experiment | Example/Notes |
|---|---|---|
| High-Quality Assembled Contigs | The substrate for gene prediction. Accuracy depends heavily on assembly quality. | Output from SPAdes, MEGAHIT, Flye. |
| Prodigal Software (v2.6.3+) | Performs fast, ab initio prokaryotic gene prediction. | Run with -p meta for metagenomes. |
| MetaGeneMark Software (v3.38+) | Gene prediction using HMMs trained on metagenomic sequences. | Requires license file for model download. |
| CD-HIT Suite (v4.8.1+) | Clusters redundant protein sequences to create a non-redundant catalog. | Essential for consensus workflows. |
| Conda/Bioconda | Reproducible environment management for installing bioinformatics tools. | environments.yaml files define tool versions. |
| Snakemake (v7.0+) | Workflow management system to orchestrate, parallelize, and track gene prediction steps. | Ensures reproducibility and scalability. |
| High-Performance Computing (HPC) Cluster or Cloud Instance | Provides computational resources for processing large metagenomic datasets. | Required for large-scale enzyme discovery projects. |
Application Notes
Within the thesis framework "A Scalable Snakemake Workflow for Targeted Enzyme Discovery in Metagenomic Datasets," Rule 4 represents the critical functional annotation layer. It transitions from gene calls (Rule 3) to biologically meaningful predictions. This rule executes two complementary homology search strategies to infer protein function, each with distinct strengths in sensitivity and specificity for enzyme discovery.
DIAMOND (BLAST-based Search): Utilized for high-speed, sensitive alignment against large, comprehensive reference databases (e.g., UniRef90, NCBI-nr). It provides broad functional labels (e.g., EC numbers, Pfam domains) and is excellent for detecting distant, but linear, sequence homology. It is the first pass for general annotation.
HMMER (Profile HMM Search): Used for high-specificity searches against custom-built Hidden Markov Model (HMM) profiles. These profiles are constructed from multiple sequence alignments of known enzyme families of interest (e.g., polyketide synthases, nitrilases). This step is crucial for targeted discovery, as it can detect remote homologs that share a common structural fold but may have low pairwise sequence identity, which DIAMOND might miss.
Integrating both methods in a single Snakemake rule ensures a balanced annotation approach: broad sensitivity (DIAMOND) coupled with targeted, family-specific precision (HMMER). The output is a consolidated annotation table, essential for downstream rules that perform filtering, prioritization, and phylogenetic analysis of candidate enzymes.
Table 1: Comparative Analysis of Homology Search Tools in Rule 4
| Feature | DIAMOND (BLASTX) | HMMER (hmmscan) | Purpose in Workflow |
|---|---|---|---|
| Search Type | Pairwise sequence alignment | Profile Hidden Markov Model alignment | Broad vs. targeted homology |
| Primary Database | UniRef90, NCBI-nr (large, flat) | Custom HMM profiles (focused, curated) | General annotation vs. family-specific discovery |
| Speed | Very Fast (~20,000x BLAST) | Moderate to Slow | Enable high-throughput screening |
| Sensitivity | High for linear homology | Very High for structural/remote homology | Catch divergent enzyme variants |
| Key Output | Best-hit taxonomic & functional ID | Bit scores, E-values, domain architecture | Candidate scoring and ranking |
Experimental Protocols
Protocol 1: DIAMOND-based Functional Annotation
Objective: To rapidly annotate predicted protein sequences with putative functions from a comprehensive reference database.
- Input: FASTA file of predicted amino acid sequences (
proteins.faa) from Rule 3 (Gene Prediction). - Database Preparation:
- Download the latest UniRef90 database:
wget ftp://ftp.uniprot.org/pub/databases/uniprot/uniref/uniref90/uniref90.fasta.gz - Decompress:
gunzip uniref90.fasta.gz - Format for DIAMOND:
diamond makedb --in uniref90.fasta -d uniref90
- Download the latest UniRef90 database:
Execution:
Run DIAMOND in
blastxmode (if starting from nucleotide contigs) orblastpmode (for protein input):Parameters:
--evalue 1e-5sets significance threshold;--max-target-seqs 5reports top 5 hits.
- Output Parsing: The tab-separated results (
diamond_results.tsv) are parsed within the Snakemake rule to extract best-hit annotations, including enzyme commission (EC) numbers from thestitlefield.
Protocol 2: Custom HMM Profile Construction & Search
Objective: To identify sequences belonging to a specific enzyme family of interest using sensitive profile HMMs.
- Input: Seed multiple sequence alignment (MSA) of a known enzyme family (e.g., from Pfam or manually curated literature data).
- HMM Profile Building:
- Align seed sequences using MUSCLE or MAFFT:
mafft --auto seed_sequences.fa > family_alignment.sto - Convert to Stockholm format if needed.
- Build the HMM profile:
hmmbuild family_profile.hmm family_alignment.sto - Calibrate the profile (for statistical scoring):
hmmpress family_profile.hmm
- Align seed sequences using MUSCLE or MAFFT:
HMMER Search:
Execute
hmmscanagainst the predicted proteome:The
--cut_gaoption uses curated model-specific thresholds for optimal family membership prediction.
- Output Interpretation: The domain table output (
hmmer_results.domtblout) lists sequences that significantly match the profile. Sequences with an independent E-value < 0.01 are considered strong candidates for the target enzyme family.
Visualizations
Diagram 1: Rule 4 Workflow in Snakemake Pipeline (90 chars)
Diagram 2: Homology Search Logic & Integration (95 chars)
The Scientist's Toolkit
Table 2: Key Research Reagent Solutions for Functional Annotation
| Item | Function in Protocol | Example/Supplier |
|---|---|---|
| UniRef90 Database | Comprehensive, clustered non-redundant protein sequence database used as the reference for DIAMOND searches. Provides standardized functional annotations. | UniProt Consortium |
| Custom HMM Profile | A statistical model representing the consensus and variation of a specific enzyme family, enabling sensitive detection of remote homologs. | Built via hmmbuild from curated alignments. |
| Pfam Database | Repository of protein family HMMs. Source of seed alignments for building or validating custom profiles for common enzyme families. | EMBL-EBI |
| MUSCLE/MAFFT Software | Tools for generating the multiple sequence alignments (MSAs) required as input for building robust and accurate HMM profiles. | EMBL-EBI, multiple sources |
| Snakemake Workflow Manager | Orchestrates the execution of DIAMOND and HMMER rules, manages dependencies, and ensures reproducible annotation across compute environments. | Open Source |
| High-Performance Compute (HPC) Cluster | Essential for parallel execution of computationally intensive homology searches against large metagenomic datasets in a feasible time. | Institutional or cloud-based (AWS, GCP). |
Within the context of a targeted enzyme discovery Snakemake workflow, Rule 5 represents the critical bioinformatic and analytical juncture where putative enzyme candidates are refined from a broad list into a prioritized subset for experimental validation. This rule integrates multi-parametric data—including sequence homology, predicted physicochemical properties, structural models, and docking scores—to filter out poor candidates, rank the remainder based on customizable criteria, and generate comprehensive, reproducible reports for research teams.
Core Components and Quantitative Metrics
The filtering and ranking process relies on defined thresholds across several computational domains. The following table summarizes typical quantitative metrics and their associated thresholds used in contemporary enzyme discovery pipelines.
Table 1: Standard Filtering Metrics and Thresholds for Enzyme Candidates
| Metric Category | Specific Metric | Typical Threshold (Inclusive) | Rationale |
|---|---|---|---|
| Sequence & Homology | Percentage Identity to Known Template | 20% - 95% (context-dependent) | Balances novelty with modeling reliability. |
| E-value (BLAST/HMMER) | < 1e-5 | Ensures significant homology. | |
| Query Coverage | > 70% | Ensures alignment spans protein of interest. | |
| Physicochemical | Predicted Solubility (Aggregation Score) | < 0.5 (lower is better) | Favors candidates likely to express solubly. |
| Instability Index | < 40 (stable) | Prioritizes thermodynamically stable proteins. | |
| pI (Isoelectric Point) | Project-specific (e.g., 5.0-9.0) | Matches downstream purification (e.g., IMAC). | |
| Structural | Model Confidence (pLDDT from AlphaFold2) | > 70 (per-residue in active site) | Ensures reliable active site geometry. |
| Ramachandran Outliers (%) | < 2% | Validates backbone torsion plausibility. | |
| Clash Score | < 10 | Indicates minimal steric conflicts. | |
| Functional | Docking Score (Binding Affinity) | Project-specific (lower ΔG better) | Ranks predicted substrate binding strength. |
| Active Site Residue Conservation | > 80% in catalytic residues | Maintains catalytic integrity. | |
| Operational | Sequence Length | Within 2 SD of family mean | Filters fragments or abnormal fusions. |
| Presence of Signal Peptide | Yes/No (as required) | Filters for secretion if needed. |
Experimental Protocols for Cited Validation Steps
Protocol 3.1: In Silico Solubility and Stability Prediction
- Objective: To computationally filter candidates prone to insolubility or instability.
- Materials: Protein sequence in FASTA format, computational tools (e.g., SOLpro, Aggrescan, ProtParam).
- Method:
- Submit candidate sequence to SOLpro (http://scratch.proteomics.ics.uci.edu/) for solubility prediction. Record the probability of solubility (0-1).
- Submit the same sequence to the ProtParam tool on the ExPASy server. Calculate the instability index; proteins with an index below 40 are considered stable.
- Run sequence through Aggrescan3D (if a structural model exists) to identify aggregation-prone surface patches.
- Analysis: Apply thresholds from Table 1. Candidates failing solubility (score <0.5) or stability (index >=40) thresholds are filtered out.
Protocol 3.2: Protein Structure Modeling and Quality Assessment using ColabFold
- Objective: To generate a reliable 3D model for active site analysis and docking.
- Materials: Multiple Sequence Alignment (MSA) of candidate, ColabFold (AlphaFold2 implementation) notebook.
- Method:
- Access the ColabFold notebook via GitHub.
- Input the candidate protein sequence(s). Use default settings for MSA generation (MMseqs2) and structure modeling.
- Execute the notebook. The output includes a predicted model file (.pdb) and a per-residue confidence metric (pLDDT).
- Validate the model using SAVES v6.0 (https://saves.mbi.ucla.edu/). Upload the .pdb file and run PROCHECK (Ramachandran plot) and ERRAT.
- Analysis: Retain models where the pLDDT score for the annotated active site residues is >70 and Ramachandran outliers are <2%. Models with poor quality scores are rejected.
Protocol 3.3: Molecular Docking for Substrate Affinity Ranking
- Objective: To rank filtered candidates based on predicted binding affinity to the target substrate.
- Materials: Protein model (.pdb), substrate molecular file (.mol2 or .sdf), docking software (e.g., AutoDock Vina, GNINA).
- Method:
- Prepare the protein: remove water, add polar hydrogens, assign Kollman charges (using tools like MGLTools for AutoDock).
- Prepare the ligand: define root and torsion trees, optimize 3D geometry.
- Define the docking search space (grid box) centered on the predicted active site.
- Execute docking with AutoDock Vina (command:
vina --receptor protein.pdbqt --ligand ligand.pdbqt --center_x y z --size_x y z --out results.pdbqt). - Extract the binding affinity (ΔG in kcal/mol) from the output log for the top pose.
- Analysis: Sort candidates by docking score (more negative ΔG indicates stronger binding). This score is a primary metric for final ranking.
Visualizations
Diagram 1: Candidate Filtering & Ranking Workflow
Diagram 2: Multi-Parametric Scoring System for Ranking
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents & Tools for Experimental Validation of Ranked Candidates
| Item | Supplier Examples | Function in Validation Pipeline |
|---|---|---|
| pET Expression Vectors | Novagen (Merck), Addgene | High-copy, T7-promoter based plasmids for recombinant protein expression in E. coli. |
| E. coli Expression Strains (BL21(DE3)) | New England Biolabs, Invitrogen | Chemically competent cells with T7 RNA polymerase gene for induced expression of pET constructs. |
| Ni-NTA Agarose Resin | Qiagen, Cytiva | Immobilized metal-affinity chromatography (IMAC) resin for purifying His-tagged recombinant enzymes. |
| Protease Inhibitor Cocktail (EDTA-free) | Roche, Thermo Scientific | Prevents proteolytic degradation of target enzymes during cell lysis and purification. |
| Size-Exclusion Chromatography (SEC) Column (HiLoad 16/600 Superdex 200 pg) | Cytiva | For final polishing step to obtain monodisperse, pure enzyme sample for kinetic assays. |
| Spectrophotometric Enzyme Assay Kit (e.g., NAD(P)H-coupled) | Sigma-Aldrich, Cayman Chemical | Enables quantitative measurement of enzyme activity and initial kinetic parameters (Km, Vmax). |
| Thermal Shift Dye (e.g., SYPRO Orange) | Invitrogen | Used in Thermofluor assays to measure protein thermal stability (Tm) under different conditions. |
| Crystallization Screening Kits (JCSG+, MORPHEUS) | Molecular Dimensions | Sparse matrix screens for identifying initial conditions for protein crystallization. |
Within the thesis framework "A Scalable Snakemake Workflow for High-Throughput Targeted Enzyme Discovery," the management of computational parameters is critical for reproducibility and adaptability. This Application Note details the implementation of centralized configuration files (YAML/JSON) to decouple experimental parameters from workflow logic, enabling rapid retargeting of the discovery pipeline to new enzyme families or organisms without code modification.
Core Configuration Architecture
The Snakemake workflow uses a hierarchical configuration system. The central config.yaml file defines all adjustable parameters, which are ingested into the Snakefile via the configfile: directive.
Table 1: Primary Configuration File Sections and Parameters
| Section | Key Parameter | Data Type | Default Value | Function in Targeted Discovery |
|---|---|---|---|---|
input |
reference_proteome |
string (file path) | null |
FASTA file of the target organism's proteome. |
known_enzyme_seeds |
list (file paths) | [] |
Curated seed sequences for the enzyme family of interest. | |
search |
hmmer_evalue |
float | 1e-5 |
E-value cutoff for HMMER homology searches. |
blastp_identity |
integer | 30 |
Minimum percent identity for BLASTp validation. | |
clustering |
cdhit_identity |
float | 0.9 |
Sequence identity threshold for CD-HIT redundancy reduction. |
output |
results_dir |
string | "results/" |
Central directory for all output files. |
Protocol: Adapting the Workflow to a New Enzyme Target
Objective: Reconfigure the existing enzyme discovery pipeline to identify novel Polyketide Synthase (PKS) domains in a newly sequenced Streptomyces genome.
Materials & Software:
- Snakemake workflow (v7.0+)
- Central
config.yamlfile - Target genome proteome (FASTA)
- Seed sequences for PKS Ketosynthase (KS) domain (PF00109)
Procedure:
- Preparation of Input Files:
a. Download the proteome of Streptomyces sp. (e.g., from NCBI RefSeq) and save as
data/streptomyces_proteome.faa. b. Obtain seed sequences. Curate a multiple sequence alignment of known PKS KS domains or download the Pfam HMM (PF00109). Save asresources/pks_ks.hmm.
Configuration File Update: a. Open the central
config.yamlfile in a text editor. b. Modify theinputsection:c. Adjust search parameters for the target family (PKS domains are well-conserved):
d. Update the
outputdirectory to reflect the experiment:Workflow Execution: a. Execute the Snakemake workflow from the command line. The workflow automatically reads the updated configuration.
b. The pipeline will run the HMMER search with the new seeds against the new proteome using the specified parameters, followed by downstream clustering and annotation steps as defined in the unchanged Snakefile.
Validation: a. Manually inspect the top hits in the final
results/streptomyces_pks_ks/candidate_hits.csv. b. Perform a conserved domain analysis (e.g., using NCBI CDD) on several candidates to confirm the presence of the PKS KS domain.
Diagram: Configuration-Driven Workflow Logic
Title: Snakemake Configuration Flow
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Computational Reagents for Targeted Discovery
| Item | Function in Configuration-Driven Workflow | Example/Format |
|---|---|---|
| YAML Configuration File | Central repository for all modifiable parameters (paths, cutoffs, thresholds). Enables adaptation without altering core code. | config.yaml |
| Reference Proteome | The set of all protein sequences from the target organism. The primary search space for enzyme discovery. | FASTA file (.faa, .fasta) |
| Seed Sequence Profile HMM | A statistical model of the enzyme family of interest, built from a curated multiple sequence alignment. Used for sensitive homology search. | HMMER3 file (.hmm) |
| Conda Environment File | Defines the exact software and versions (HMMER, BLAST+, CD-HIT) to ensure computational reproducibility. | environment.yaml |
| Sample Sheet | A CSV file listing biological sample IDs and metadata. Can be referenced in the config file for batch processing. | CSV file |
Application Notes
Effective deployment of the Snakemake workflow for targeted enzyme discovery is critical for scaling computational research from initial validation to high-throughput screening. The transition from local execution to cloud environments enables access to vast, elastic compute resources necessary for processing large genomic and metagenomic datasets.
Local Cluster Deployment: Utilizes on-premise High-Performance Computing (HPC) or institutional clusters (e.g., using SLURM, SGE). This is ideal for controlled, medium-scale analyses where data governance is strict. Performance is bounded by local infrastructure.
Cloud Deployment (AWS, GCP): Provides scalable, on-demand resources. AWS Batch and Google Cloud Life Sciences are native services for orchestrating workflow jobs. Costs are variable and depend on instance selection, storage, and data egress. Cloud deployment is essential for reproducing large-scale analyses and accessing specialized machine types (e.g., memory-optimized for assembly).
Quantitative Comparison of Deployment Environments:
| Feature / Metric | Local Laptop | Local HPC Cluster | AWS Cloud | Google Cloud Platform |
|---|---|---|---|---|
| Typical Setup Time | Minutes (local install) | 1-5 days (account/project) | 1-2 hours (config, IAM) | 1-2 hours (config, IAM) |
| Max Scalability Limit | 1 machine | 100s of nodes (fixed) | 1000s of nodes (elastic) | 1000s of nodes (elastic) |
| Cost Model | Fixed hardware cost | Institutional allocation | Pay-per-use ($/vCPU-hr) | Pay-per-use ($/vCPU-hr) |
| Approx. Cost per 1000 vCPU-hr | N/A | N/A (allocated) | ~$80 - $240 (varies by instance) | ~$70 - $220 (varies by instance) |
| Data Egress Cost | N/A | N/A | ~$0.09/GB | ~$0.12/GB |
| Optimal Use Case | Rule development, debugging | Routine scheduled analyses | Burst, large-scale, reproducible runs | Burst, large-scale, integrated services (BigQuery) |
| Snakemake Integration | --cores |
--cluster (SLURM, etc.) |
--kubernetes or AWS Batch plugin |
--kubernetes or GCP Life Sciences plugin |
Experimental Protocols
Protocol 1: Deploying Snakemake on a Local SLURM Cluster
Objective: Execute the enzyme discovery workflow on an institutional HPC cluster.
- Environment Setup: On the cluster login node, install Miniconda. Create a conda environment using the
environment.ymlfile exported from the development setup. - Profile Configuration: Create a Snakemake profile directory for SLURM. Configure a
config.yamlfile within it:
- Data Staging: Transfer input genomic files to the cluster's shared filesystem (e.g., Lustre, NFS).
- Workflow Submission: Execute the workflow using the profile:
snakemake --profile /path/to/slurm_profile --use-conda. - Monitoring: Monitor job status via
squeueand Snakemake's output. Final results will be in the designated output directory.
Protocol 2: Deploying Snakemake on AWS using AWS Batch
Objective: Scale enzyme discovery workflow elastically using AWS.
- Prerequisites: AWS CLI configured, an S3 bucket for input/output data.
- Infrastructure Setup: Use the
snakemake-executor-plugin-aws-batchTerraform template to create a VPC, an S3-accessible EFS filesystem, a Batch compute environment, job queue, and job definition. - Data Transfer: Upload all input files (
seqs/*.fastq,databases/*.hmm) from the local system to the S3 bucket:aws s3 sync input/ s3://<bucket-id>/input/. - Configuration: Create a Snakemake config file (
config/config_aws.yaml):
- Execution: Run the workflow:
snakemake --executor aws_batch --default-remote-provider S3 --default-remote-prefix <bucket-id>/snakemake --use-conda -c 1. The-c 1limits the local driver to one thread. - Monitoring & Download: Monitor jobs in the AWS Batch console. Upon completion, download results:
aws s3 sync s3://<bucket-id>/snakemake/output/ ./results/.
Protocol 3: Deploying Snakemake on GCP using Google Cloud Life Sciences
Objective: Execute workflow on GCP, leveraging integration with other Google services.
- Prerequisites:
gcloudCLI installed and initialized. A Google Cloud Storage (GCS) bucket created. - Service Enablement: Enable the Cloud Life Sciences, Compute Engine, and GCS APIs.
- Data Transfer: Upload input data to GCS:
gsutil -m cp -r input/*.fastq gs://<bucket-id>/input/. - Configuration: Create a Snakemake profile for GCP Life Sciences. Key settings in
config.yaml:
- Execution: Run from a local machine or a Cloud Shell:
snakemake --profile ./gcp_profile --default-remote-prefix gs://<bucket-id>/snakemake. - Post-processing: Results will be written to the GCS bucket. Use
gsutilfor download or connect results to BigQuery for downstream analysis.
Visualization
Diagram 1: Snakemake Deployment Pathway for Enzyme Discovery
Diagram 2: AWS Batch Execution Architecture for Snakemake
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Deployment Context |
|---|---|
| Snakemake Workflow Management System | Core tool for defining, executing, and scaling the reproducible bioinformatics workflow for enzyme discovery. |
| Conda / Bioconda / Mamba | Provides isolated, reproducible software environments for each workflow rule, ensuring consistent tool versions across all deployments. |
| Docker / Singularity Containers | Containerization solutions to package entire analysis environments, guaranteeing portability between local, cluster, and cloud execution. |
| AWS CLI / gcloud CLI | Command-line tools for configuring, managing, and transferring data to and from AWS and GCP cloud resources, respectively. |
| Terraform / CloudFormation | Infrastructure-as-Code (IaC) tools to programmatically provision and manage the cloud infrastructure (VPC, compute, storage) needed for workflow execution. |
| S3 / Google Cloud Storage (GCS) | Scalable, durable object storage services used as the primary remote data layer for input, output, and intermediate files in cloud deployments. |
| SLURM / SGE Scheduler | Job scheduling and resource management software used to efficiently execute workflow jobs on local HPC clusters. |
| Git / GitHub / GitLab | Version control systems essential for tracking changes to the Snakefile, configuration profiles, and analysis scripts, enabling collaboration and reproducibility. |
Streamlining Your Search: Debugging, Scaling, and Cost-Effective Optimization
Common Snakemake Errors and Debugging Strategies in Metagenomic Contexts
This document serves as an application note within a broader thesis on developing a scalable Snakemake workflow for targeted enzyme discovery from complex metagenomic datasets. It outlines prevalent errors and systematic debugging approaches to ensure robust, reproducible bioinformatics analyses.
Common Snakemake Errors: Identification and Resolution
The table below summarizes frequent errors, their typical causes in metagenomic analyses, and immediate resolution strategies.
| Error Category | Specific Error Message/ Symptom | Common Cause in Metagenomics | Immediate Debugging Action |
|---|---|---|---|
| Rule Dependency | MissingInputException |
Incorrect wildcard pattern matching for highly variable sample names (e.g., sample_001_R1.fq vs sample001_R1.fastq). |
Run snakemake -n -p to visualize the DAG and check expected vs. actual filenames. Use wildcard_constraints in rules. |
| Resource Deadlock | WorkflowError: Cyclic dependency |
Circular logic in rule definitions, common when post-assembly analysis (e.g., binning) is incorrectly fed back into assembly. | Generate a rulegraph (snakemake --rulegraph). Inspect cycles and restructure workflow into a linear DAG. |
| Cluster/Cloud Execution | Job failed with exit code 1 outside of rule. |
Insufficient memory (mem) or runtime (time) defined for resource-intensive steps like metaSPAdes assembly or read mapping. |
Check cluster logs. Implement resource scaling using profiles (e.g., --profile slurm) and define resources: in rules. |
| Configuration Errors | KeyError in config dict. |
Referencing a undefined sample key or path in the config.yaml file, especially with large sample batches. | Validate config file with a YAML linter. Use config.get("key", default) for optional parameters in scripts. |
| Permission & Paths | FileNotFoundError or Permission denied |
Relative vs. absolute path issues when using working directory (--directory) or containerized execution (--use-singularity). |
Use absolute paths in config.yaml. Prepend workflow.basedir to relative paths in rules. Check file permissions. |
| Software Environment | ModuleNotFoundError in Python script. |
Inconsistent Conda environments across rules or missing specific dependency versions for tools like DIAMOND or Prokka. | Use conda: directives per rule. Run snakemake --use-conda --conda-cleanup-pkgs. Isolate tool environments. |
| Ambiguous Wildcards | WildcardError in rule all. |
Multiple rules can produce the same output file extension (e.g., .txt), confusing the final target rule. |
Make output filenames more specific (e.g., gene_count.txt vs sample_stats.txt). Order rules precisely. |
Debugging Protocol: A Stepwise Methodology
Protocol 1: Systematic Snakemake Workflow Debugging
Objective: To diagnose and resolve failures in a metagenomic Snakemake workflow, from quality control through to gene annotation and abundance profiling.
Materials:
- A functional Snakemake installation (v7.0+).
- Workflow Snakefile and configuration file.
- Sample metagenomic read files (e.g., FASTQ format).
- Access to a high-performance computing (HPC) cluster or local server with sufficient resources.
Procedure:
- Dry-run and DAG Generation:
- Execute
snakemake -n -p. This performs a dry-run (-n) while printing shell commands (-p), verifying the workflow logic without execution. - Generate a directed acyclic graph (DAG) visualization:
snakemake --dag | dot -Tsvg > workflow_dag.svg. Visually inspect for missing edges or unexpected nodes.
- Execute
Rulegraph Analysis for Structure:
- Generate a rulegraph to understand high-level rule dependencies:
snakemake --rulegraph | dot -Tsvg > workflow_rulegraph.svg. This abstract view helps identify cyclic dependencies.
- Generate a rulegraph to understand high-level rule dependencies:
Targeted Execution with Debug Flags:
- If a specific rule fails, run Snakemake targeting its output with verbose debugging:
snakemake <target_file> -r -n. The-rflag prints the reason for executing each job. - Use
--debug-dagfor detailed debugging information on dependency resolution.
- If a specific rule fails, run Snakemake targeting its output with verbose debugging:
Log File Inspection:
- Every rule should direct
output,log, anderrorstreams to distinct files (e.g.,log: "logs/{sample}.trimmomatic.log"). - Examine the
logand cluster error files (slurm-*.out) for application-specific errors (e.g.,trimmomaticadapter file not found,diamonddatabase corruption).
- Every rule should direct
Environment and Resource Validation:
- For Conda errors, recreate the environment manually:
conda env create -f <rule_env.yaml>and test the shell command. - For cluster job failures, profile resource usage (CPU, memory) of the failing tool on a subset of data and update the rule's
resources:section accordingly.
- For Conda errors, recreate the environment manually:
Config File Validation:
- Use a Python script to load and print the config:
python -c "import yaml; print(yaml.safe_load(open('config.yaml')))". Ensure all sample keys and paths are correct.
- Use a Python script to load and print the config:
Visual Workflow and Error Mapping
Diagram 1: Metagenomic Snakemake Workflow DAG
Diagram 2: Snakemake Error Debugging Decision Tree
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Metagenomic Snakemake Workflow |
|---|---|
| Conda/Mamba | Creates isolated, reproducible software environments for each bioinformatics tool (e.g., specific versions of SPAdes, DIAMOND), preventing dependency conflicts. |
| Snakemake Profiles | Pre-configured sets of command-line options (e.g., for SLURM, AWS) that manage cluster submission parameters (queue, memory, time) without modifying the Snakefile. |
| Config.yaml File | Centralizes all sample metadata, file paths, database locations, and tool parameters, enabling workflow adaptation to new projects by editing a single file. |
| Singularity/Apptainer | Containerization technology that packages entire tool environments, guaranteeing absolute computational reproducibility across different HPC systems. |
| Standardized Logging | Directive (log:) in each rule that captures stdout and stderr to dedicated files, providing an audit trail for debugging and reproducibility. |
| Cluster Job Manager | (e.g., SLURM, SGE). Manages resource allocation and job scheduling on HPC systems, integrated seamlessly via Snakemake's --cluster or profiles. |
| Git | Version control for the Snakefile, configuration, and analysis scripts, tracking changes and enabling collaboration on the workflow itself. |
| MultiQC | Aggregates results from multiple tools (FASTQC, bowtie2, etc.) into a single interactive HTML report, providing a unified overview of the entire analysis run. |
Within a broader thesis on developing a Snakemake workflow for targeted enzyme discovery, efficient resource management is paramount. High-throughput virtual screening, molecular dynamics simulations, and genomic analyses demand strategic allocation of computational resources to balance runtime, memory footprint, and cloud/CPU costs. This document provides application notes and protocols for optimizing these factors within a reproducible workflow framework.
Quantitative Data on Resource Trade-offs
Table 1: Comparative Resource Profiles for Common Enzyme Discovery Tasks
| Task | Typical Runtime | Peak Memory (GB) | Estimated Cost (Cloud, per run) | Primary Scaling Factor |
|---|---|---|---|---|
| Homology Modeling (Modeller) | 2-10 hours | 8-16 | $0.40 - $2.00 | Target sequence length |
| Molecular Docking (AutoDock Vina) | 5-30 min/ligand | 2-4 | $0.05 - $0.30/ligand | Ligand library size |
| MD Simulation (GROMACS, 100ns) | 24-72 hours | 32-128 | $15 - $60 | System size, simulation time |
| Metagenomic Assembly (MEGAHIT) | 4-12 hours | 64-256 | $5 - $25 | Sequencing depth (GB) |
| HMMER Sequence Search | 1-6 hours | 4-8 | $0.20 - $1.50 | Database & query size |
Experimental Protocols
Protocol 1: Configurable Snakemake Workflow for Batch Docking
Objective: To perform virtual screening of a compound library against an enzyme target with tunable resource parameters.
- Input Preparation:
- Prepare the receptor protein file in PDBQT format (using
prepare_receptorfrom AutoDockTools). - Prepare ligand library in SDF, convert to PDBQT using
obabelor a custom script within a Snakemake rule.
- Prepare the receptor protein file in PDBQT format (using
- Snakemake Rule Design:
- Create a rule
run_dockingthat takes a single ligand PDBQT as input. - In the
resources:directive of the rule, definemem_mb,runtime, and a customcostparameter. - Use
threads:to leverage multi-core support in Vina (e.g.,--cpu 8).
- Create a rule
- Resource-Aware Execution:
- Use a profile (e.g., for SLURM, AWS Batch) to link Snakemake resource directives to cluster/cloud scheduler.
- Implement checkpointing: If a job fails due to time/memory, the rule's
resourcesare automatically doubled on retry.
- Aggregation: Use a final rule to collate all output DLG/CSV files into a single ranked results table.
Protocol 2: Memory-Efficient Metagenomic Gene Calling
Objective: To identify putative enzyme-encoding genes from large metagenomic assemblies without exhausting RAM.
- Quality Control & Assembly: (Refer to Table 1 for resources). Perform trimming (Trimmomatic) and assembly (MEGAHIT with
--mem-flagset to control memory use). - Gene Prediction: Use
Prodigalin meta-mode (-p meta). It is memory-efficient and streams the assembly.- Critical Step: Do not load the entire assembly file into memory. Process via streaming using Snakemake's
pipe()command or by chunking the input in a shell directive.
- Critical Step: Do not load the entire assembly file into memory. Process via streaming using Snakemake's
- HMMER Search with
--chunk: For searching predicted genes against Pfam HMMs:- Split the protein FASTA into chunks (e.g., 10,000 sequences per chunk).
- Create a Snakemake rule that processes each chunk (
rule hmmsearch_chunk), specifyingmem_mb=8192. - Use HMMER's
--cpuflag and setthreads:accordingly.
- Results Merging: A final rule concatenates all chunk results using
hmmsearch's table output option (--tblout).
Visualizations
Title: Snakemake Resource Optimization Logic Flow
Title: Core Resource Trade-off Relationships
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools for Resource-Optimized Enzyme Discovery
| Tool/Solution | Function in Workflow | Key Resource Consideration |
|---|---|---|
| Snakemake | Workflow management. | Defines resources: directives (mem, runtime) for each rule, enabling efficient scheduling and cost tracking. |
| Conda/Mamba | Environment management. | Ensures reproducible software installations, avoiding conflicts and version-related inefficiencies. |
| Docker/Singularity | Containerization. | Guarantees portability across HPC/cloud, standardizing runtime environment and performance. |
| GROMACS | Molecular dynamics. | Highly optimized for CPU parallelization; scaling efficiency drops beyond optimal core count. |
| AutoDock Vina/QuickVina 2 | Molecular docking. | Embarrassingly parallel per ligand; ideal for batch arrays with low inter-task communication. |
| HMMER3 | Sequence homology search. | Can be chunked (--chunk) to process large databases in fixed memory segments. |
| Prodigal | Gene prediction. | Streams input (low memory) and is fast, ideal for large metagenomic contigs. |
| Slurm/AWS Batch Profile | Cluster/cloud job scheduling. | Translates Snakemake resource requests into native scheduler commands for precise allocation. |
Within the broader thesis developing a Snakemake workflow for targeted enzyme discovery, managing the scale and complexity of metagenomic data is a primary bottleneck. This protocol details the efficient data management and I/O strategies essential for processing terabytes of raw sequencing reads through quality control, assembly, gene prediction, and functional annotation within the reproducible Snakemake framework, ultimately enabling high-throughput screening for novel biocatalysts.
Core I/O Challenges and Benchmarking Data
Efficient workflow design requires understanding performance trade-offs. The following table summarizes key benchmarks for common data operations on a high-performance computing (HPC) cluster with a parallel file system (e.g., Lustre, BeeGFS).
Table 1: I/O Performance Benchmarks for Common Metagenomic Data Operations
| Data Operation | Typical Data Volume | Recommended Tool/Format | Approx. Time (Example) | Critical I/O Factor |
|---|---|---|---|---|
| Raw Read Transfer (from sequencer) | 500 GB - 2 TB per lane | rsync, md5sum validation |
1-4 hours (10 GbE network) | Network bandwidth, integrity checks |
| Compressed FASTQ Storage | Compression ratio: ~0.3 (70% reduction) | gzip (.fq.gz), bioinformatics-optimized pbzip2 |
N/A | Storage footprint, decompression speed |
| Multi-sample Quality Control | 100 samples x 10 GB each | Snakemake parallelization, FastQC (MultiQC aggregation) |
2 hrs (parallel) vs. 20 hrs (serial) | Parallel file reads, aggregated report I/O |
| Metagenomic Assembly (Co-assembly) | 1 TB merged reads | MEGAHIT (uses succinct de Bruijn graph) |
24-72 hours CPU/GPU | Intermediate file I/O, memory-disk swapping |
| Gene Catalog Creation (Prediction) | 500 GB assembly output | Prodigal (parallel run on contigs) |
6-12 hours | High number of small file writes |
| Functional Annotation (vs. DB) | 10-100 million protein sequences | DIAMOND (blastx mode, chunked), MMseqs2 |
8-48 hours (depending on DB size) | Database indexing, streaming I/O |
| Final Results Table | ~10 GB (parquet format) | Pandas/DuckDB, Apache Parquet format |
Query: seconds | Columnar storage for fast analytics |
Application Notes & Protocols
Protocol 3.1: Hierarchical Data Organization for a Snakemake Project
Objective: Create a reproducible directory structure that separates raw data, processed intermediates, and results, optimizing for Snakemake's dependency resolution and HPC file system performance.
- Directory Structure Creation:
- Symlink Raw Data: For immutable raw data (e.g., from sequencing core), create symlinks from
data/raw/to the read-only storage location. This preserves path structure without duplication. - Sample Sheet Management: Create a CSV file (
resources/samples.csv) mapping sample IDs to file paths. This is the central input for the Snakemake workflow. - Snakemake Configuration: In
config/config.yaml, define relative paths to these directories. Use wildcards ({sample}) in rule inputs/outputs to leverage the structure.
Protocol 3.2: Implementing Checkpoint-Based Streaming for Large-Scale Assembly
Objective: Process co-assembly of hundreds of samples without creating prohibitively large intermediate FASTQ files.
- Read List Preparation: Generate a text file (
resources/read_list.txt) containing the absolute paths to all cleaned read files (e.g.,cleaned/Sample1.fq.gz,cleaned/Sample2.fq.gz). Snakemake Checkpoint Rule: Write an assembly rule that takes the
read_list.txtas input. The rule script does not concatenate files but streams them.Downstream Rule Integration: Use
checkpointfunctionality to allow subsequent gene prediction rules (prodigal) to wait for assembly completion and then dynamically discover the resulting contig files (final.contigs.fa) as their input.
Protocol 3.3: Optimized I/O for Annotation with Large Databases
Objective: Annotate 50M predicted genes against the UniRef90 database (>50 GB) efficiently.
Database Preparation:
Chunked Alignment Strategy: Implement a Snakemake rule that splits the gene catalog into chunks, processes chunks in parallel, and merges results.
I/O Tuning: Set
--tmpdirto a node-local SSD or RAM disk to avoid network file system contention for temporary files.
Mandatory Visualizations
Diagram 1: Snakemake I/O-Optimized Workflow for Enzyme Discovery
Diagram 2: Data Lifecycle & Storage Tier Strategy
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Data Management "Reagents" for Large-Scale Metagenomics
| Item | Function in Workflow | Key Consideration |
|---|---|---|
| Snakemake Workflow Manager | Orchestrates all steps, manages dependencies, and enables reproducible parallel execution. | Use --cores and --resources for HPC scheduling; checkpoint for dynamic workflows. |
Linux Core Utilities (rsync, md5sum) |
Secure data transfer and integrity verification for raw reads. | Always checksum after transfer. Use rsync -avcP. |
| Parallel File System (e.g., Lustre) | High-throughput storage for concurrent access by hundreds of workflow jobs. | Optimize stripe count for file types (many small files vs. single large file). |
Compression Tools (pigz, pbzip2) |
Multi-threaded compression/decompression to reduce storage and network I/O. | Use --processes flag. Balance compression level (-1 to -9) with CPU time. |
| Chunking Script (Custom Python/AWK) | Splits massive sequence files into smaller, parallelizable units for annotation steps. | Chunk size should be ~1-10M sequences to balance overhead and parallelism. |
| Columnar Storage Format (Apache Parquet) | Stores final annotation and abundance tables for fast querying and analysis in R/Python. | Highly compressed, enables reading subsets of columns without full file load. |
| Containers (Singularity/Apptainer) | Packages complex software (MEGAHIT, DIAMOND) and dependencies for consistent HPC execution. | Build from Dockerhub images; ensures version reproducibility. |
| Metadata Catalog (e.g., SQLite) | Lightweight database tracking sample metadata, file paths, and processing parameters. | Essential for linking experimental conditions to computational results in enzyme discovery. |
Within the overarching thesis of developing a robust Snakemake workflow for targeted enzyme discovery, the construction of high-specificity custom Hidden Markov Model (HMM) libraries represents a critical bioinformatics preprocessing step. Public databases like Pfam provide broad-spectrum models that often lack the specificity required for distinguishing between closely related enzyme subfamilies, leading to ambiguous annotations. Custom HMM libraries, built from carefully curated, phylogenetically informed seed alignments, dramatically improve the precision of homology searches in metagenomic and genomic datasets, directly impacting the downstream identification of novel biocatalysts for drug development.
Application Notes: The Rationale for Custom HMMs
Limitations of Generalist Databases
Public HMM repositories are designed for general applicability, which can result in:
- Over-prediction: Assignment of sequences to overly broad functional categories.
- Reduced Resolution: Inability to differentiate between paralogous enzyme families with divergent substrate specificities.
- Discovery Blind Spots: Failure to detect novel family clades that are distant from canonical seed sequences.
Advantages in Targeted Discovery
A custom HMM library, focused on a specific enzyme class (e.g., Glycosyl Hydrolase Family 7 or Polyketide Synthase modules), allows researchers to:
- Set Precise Boundaries: Define model thresholds based on project-specific sensitivity/specificity trade-offs.
- Incorporate Novel Diversity: Include recently characterized or proprietary sequences from internal research.
- Streamline Workflows: Integrate directly into automated Snakemake pipelines for reproducible, large-scale dataset screening.
Protocols for Building and Curating Custom HMM Libraries
Protocol 3.1: Seed Alignment Curation and Preparation
Objective: To assemble a high-quality, non-redundant, and phylogenetically representative multiple sequence alignment (MSA) as the input for HMM building.
Materials & Input Data:
- A set of trusted, full-length protein sequences representing the target enzyme family.
- Software: Clustal Omega, MAFFT, HMMER suite, CD-HIT, FastTree.
Methodology:
- Initial Sequence Collection: Gather sequences from public (UniProt, NCBI) and proprietary sources. Include experimentally characterized representatives.
- Redundancy Reduction: Use CD-HIT with a 90% identity threshold to cluster sequences and remove identical copies.
Alignment Generation: Align sequences using MAFFT for accuracy with complex indel patterns.
Alignment Trimming: Trim poorly aligned columns and termini using trimAl with a gap threshold of 70%.
Phylogenetic Assessment: Build a quick neighbor-joining tree (FastTree) to visualize clustering and identify potential outliers or mis-annotated sequences. Manually inspect and refine the seed alignment.
Data Output: A curated, trimmed MSA in STOCKHOLM or FASTA format.
Protocol 3.2: HMM Building and Calibration
Objective: To convert the curated MSA into a calibrated HMM profile.
Methodology:
- HMM Build: Use
hmmbuildfrom the HMMER suite.
Model Calibration: Calibrate the model with
hmmpressto generate statistical parameters for E-value calculation. This step is essential for accurate significance assessment during searches.Threshold Determination: Search the model against a known reference database (e.g., Swiss-Prot) using
hmmsearch. Analyze the score distribution to define gathering (GA) thresholds that optimize for family-specific inclusion.
Protocol 3.3: Integration into a Snakemake Workflow
Objective: To automate the custom HMM library creation and its application for screening.
Methodology:
- Define the workflow in a
Snakefile. Key rules include:rule get_seeds: Downloads initial seed sequences.rule build_alignment: Runs alignment and trimming.rule build_hmm: Executeshmmbuildandhmmpress.rule hmm_search: Applies the custom library to a target metagenomic protein dataset.
Example Snakefile Snippet:
Data Presentation
Table 1: Performance Comparison of Custom vs. Pfam HMM for Glycosyl Hydrolase Family 7 Screening
| Metric | Pfam Model (PF00840) | Custom GH7 Library | Notes |
|---|---|---|---|
| Number of Hits (E-value < 1e-10) | 1,245 | 892 | Custom model reduces over-calling. |
| Avg. E-value of True Positives | 4.2e-45 | 1.1e-78 | Custom model yields more significant hits for true family members. |
| Alignment Coverage (%) | 78.2 ± 12.4 | 94.7 ± 3.1 | Custom model provides more complete domain alignment. |
| Runtime (minutes) | 22.1 | 18.5 | Smaller, focused library searches faster. |
| False Positive Rate (%)* | 8.5 | 2.1 | Based on validation against characterized negative set. |
*False positives defined as hits with no detectable activity in subsequent functional screens.
Visualizations
HMM Library Curation & Integration Workflow
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Research Reagents & Tools for Custom HMM Development
| Item | Function in Protocol | Example/Supplier |
|---|---|---|
| Curated Reference Sequences | Provides the foundational seed data for model building. | UniProtKB, internal characterization data. |
| Multiple Sequence Alignment Tool | Generates the positional homology map from seed sequences. | MAFFT (v7.520), Clustal Omega. |
| Alignment Trimming Software | Removes noisy, poorly aligned columns to improve model signal. | trimAl (v1.4), BMGE. |
| HMMER Suite | Core software for building, calibrating, and searching with HMMs. | http://hmmer.org/ |
| High-Performance Computing (HPC) Cluster | Enables alignment, tree-building, and large-scale searches. | Local SLURM or SGE cluster, cloud compute (AWS, GCP). |
| Workflow Management System | Automates and reproduces the entire multi-step process. | Snakemake (v7.25+). |
| Target Protein Dataset | The query to be screened with the custom HMM library. | Metagenome-assembled genomes (MAGs), genomic databases. |
This application note details the implementation of containerization and environment management strategies to ensure computational reproducibility within a Snakemake-based pipeline for targeted enzyme discovery. This pipeline is central to a broader thesis on high-throughput metagenomic screening for novel biocatalysts. Reproducibility is a cornerstone of computational science, and the combined use of Conda for package management and Docker/Singularity for OS-level encapsulation guarantees that all software dependencies are precisely defined, versioned, and isolated, yielding identical results across different computing platforms.
Current State of Tools and Adoption
The following table summarizes key metrics and features of the core technologies, based on recent community data and documentation.
Table 1: Comparison of Reproducibility Technologies (2023-2024)
| Technology | Primary Use | Key Metric (Approx.) | Snakemake Integration | Key Advantage for Enzyme Discovery |
|---|---|---|---|---|
| Conda/Bioconda | Package & Environment Management | >10,000 bioinformatics packages available | Native (conda:) |
Vast, curated repository of bioinformatics tools (e.g., DIAMOND, HMMER, Prokka). |
| Docker | OS-level Containerization | ~13 million developers use Docker (2023) | Via --use-singularity flag |
Consistent runtime environment from laptop to cloud; ideal for development. |
| Singularity/Apptainer | HPC-friendly Containerization | Default on >70% of Top100 HPC systems (2024) | Native (container:) or --use-singularity |
Runs without root privileges, essential for shared High-Performance Computing clusters. |
| Snakemake | Workflow Management | >5,000 citations (2024) | N/A | Directives for Conda and containers unify environment specification within the workflow. |
Integrated Protocol for a Reproducible Snakemake Pipeline
This protocol outlines the step-by-step integration of Conda and Singularity within a Snakemake workflow for enzyme discovery, encompassing steps from read preprocessing to functional annotation.
Protocol 3.1: Defining Conda Environments for Pipeline Stages
Objective: Create isolated, versioned software environments for each major analysis module.
Materials:
- Workstation with Miniconda installed.
- Snakemake (≥7.0) installed via Conda.
Procedure:
- For each pipeline stage, create a separate YAML file (e.g.,
envs/quality.yaml,envs/assembly.yaml,envs/annotation.yaml). - Specify all dependencies with exact versions. Use channels
conda-forgeandbioconda. - Example:
envs/annotation.yamlfor gene calling and annotation.
Protocol 3.2: Building a Singularity Image from a Dockerfile
Objective: Create a portable Singularity container image encapsulating the entire system environment.
Materials:
- System with Docker and Singularity/Apptainer installed.
Dockerfiledefining the base OS and core utilities.
Procedure:
- Create a
Dockerfilestarting from a minimal base (e.g.,ubuntu:22.04orcontinuumio/miniconda3:24.1.0-0). - Install system libraries required by bioinformatics tools (e.g.,
zlib,bzip2). - Copy the Conda environment YAML files into the image and create the environments using
conda env create -f <file.yaml>. - Set the default
PATHto include the Conda environment binaries. - Build the Docker image:
docker build -t enzyme-pipeline:1.0 . - Convert the Docker image to a Singularity Image File (SIF):
singularity build pipeline_v1.0.sif docker://enzyme-pipeline:1.0.
Protocol 3.3: Executing the Integrated Snakemake Pipeline on an HPC Cluster
Objective: Run the complete, reproducible pipeline on a shared compute cluster.
Materials:
- HPC cluster with Singularity/Apptainer and Conda installed.
- Snakemake profile configured for the cluster scheduler (e.g., SLURM).
- Singularity SIF file (
pipeline_v1.0.sif). - Snakemake workflow files (
Snakefile,config.yaml).
Procedure:
- Structure the
Snakefileto use bothcondaandcontainerdirectives per rule. - Example Rule for HMM Search:
- Submit the workflow:
snakemake --use-conda --use-singularity --jobs 12 --profile slurm. Snakemake will:- Activate the specified Conda environment inside the Singularity container.
- Execute the rule's command in this fully encapsulated environment.
- Manage job submission to the cluster via the profile.
Visualizations
Workflow for Reproducible Enzyme Discovery
Software Stack for Reproducible Analysis
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Digital Research "Reagents" for Reproducible Enzyme Discovery
| Item | Function in Protocol | Example/Version |
|---|---|---|
| Conda Environment File (.yaml) | Defines the precise software stack and versions for a discrete analytical step, ensuring dependency resolution. | envs/assembly.yaml |
| Dockerfile | Blueprint for building a Docker image. Specifies the base operating system, system libraries, and instructions to install Conda environments. | FROM ubuntu:22.04 |
| Singularity Image File (.sif) | Immutable, portable container file executed directly on HPC systems. Contains the entire runtime environment. | pipeline_v1.0.sif |
| Snakemake Profile | Configuration file for a specific compute platform (e.g., SLURM, SGE). Manages job submission parameters (time, memory, queue). | config.yaml in ~/.config/snakemake/ |
| Conda Package Repositories | Curated channels providing pre-compiled bioinformatics software. Bioconda is essential for life sciences tools. | bioconda, conda-forge |
| Container Registries | Cloud repositories for storing and sharing built container images. Enables versioning and distribution of the runtime environment. | Docker Hub, GitHub Container Registry, Sylabs Cloud |
Monitoring Workflow Performance with Snakemake's Logging and Reporting Features
Within the targeted enzyme discovery research thesis, robust workflow monitoring is critical for reproducibility and performance optimization. Snakemake's integrated logging, reporting, and profiling features provide essential mechanisms for tracking pipeline execution, debugging failures, and quantifying resource utilization across large-scale bioinformatics analyses. This document details protocols for implementing comprehensive performance monitoring in enzyme discovery workflows, enabling researchers to identify bottlenecks in genome annotation, sequence alignment, and structural prediction steps.
Targeted enzyme discovery workflows involve computationally intensive steps: genomic database mining, multiple sequence alignment, phylogenetic analysis, homology modeling, and functional annotation. Snakemake's monitoring tools allow researchers to track each step's success, execution time, and computational load, ensuring efficient resource allocation in high-performance computing (HPC) environments.
Core Monitoring Features: Protocols and Implementation
Directed Acyclic Graph (DAG) Visualization Protocol
Purpose: Visualize workflow structure and dependencies before execution. Protocol:
- Generate the DAG representation in DOT format:
- For targeted enzyme workflows, annotate key rule groups:
Table 1: DAG Generation Parameters
| Parameter | Value | Description |
|---|---|---|
--dag |
N/A | Generates DOT format graph |
-Tsvg |
SVG output | Vector graphic format |
-Tpng |
PNG output | Raster graphic format |
| Resolution | 300 dpi | Publication quality |
Execution Logging Protocol
Purpose: Capture runtime information, errors, and standard output/error streams. Protocol:
Table 2: Log File Management
| Log Type | Location | Retention Policy | Critical Information |
|---|---|---|---|
| Rule execution | logs/rule_name.log |
90 days | Parameters, start/end times |
| Workflow summary | logs/snakemake.log |
Permanent | Overall success/failure |
| Cluster submission | cluster/submission_logs/ |
30 days | Job IDs, queue status |
Performance Profiling Protocol
Purpose: Measure computational resource utilization across workflow steps. Protocol:
- Enable benchmarking for critical rules:
- Generate comparative analysis:
Table 3: Benchmark Metrics Collected
| Metric | Unit | Tool | Typical Range (Enzyme Discovery) |
|---|---|---|---|
| CPU time | seconds | Snakemake | 60-10,000 |
| Wall clock time | seconds | Snakemake | 30-8,000 |
| Memory usage | MB | Snakemake | 500-32,000 |
| I/O read | MB | Snakemake | 10-5,000 |
| I/O write | MB | Snakemake | 1-1,000 |
HTML Reporting Protocol
Purpose: Generate interactive execution reports for collaboration and publication. Protocol:
- Execute workflow with reporting flag:
- Customize report sections in configuration:
Table 4: HTML Report Components
| Component | Data Source | Visualization |
|---|---|---|
| Rule graph | DAG | Interactive SVG |
| Timeline | Benchmark data | Gantt chart |
| Statistics | Log files | Summary tables |
| File sizes | Output files | Tree map |
| Parameters | Config files | JSON viewer |
Advanced Monitoring: Cluster Integration
HPC/Slurm Monitoring Protocol
Purpose: Track workflow performance on cluster environments.
Real-time Dashboard Protocol
Purpose: Monitor active workflow execution.
Visualization of Monitoring Workflow
Diagram Title: Snakemake Monitoring Workflow Architecture
The Scientist's Toolkit: Research Reagent Solutions
Table 5: Essential Monitoring and Analysis Tools
| Item | Function | Specification |
|---|---|---|
| Snakemake v7+ | Workflow management | Required for reporting features |
| Graphviz | DAG visualization | ≥2.40 for SVG export |
| Python pandas | Benchmark analysis | ≥1.4 for data manipulation |
| R ggplot2 | Performance plotting | ≥3.3 for publication figures |
| Jupyter Lab | Interactive analysis | ≥3.0 for report development |
| SLURM manager | Cluster monitoring | ≥20.11 for HPC integration |
| Prometheus | Resource metrics | ≥2.30 for time-series data |
| Grafana | Monitoring dashboard | ≥8.0 for visualization |
Experimental Protocol: Comprehensive Workflow Audit
Protocol Title: Monthly Performance Audit for Enzyme Discovery Pipeline
Materials:
- Snakemake workflow with benchmark directives
- HPC cluster access with Slurm
- 1 TB storage for log retention
- PostgreSQL database for metrics (optional)
Procedure:
- Baseline Execution: Run complete workflow with
--benchmark-repeats 5 - Log Aggregation: Collect all rule logs to central directory
- Metric Extraction: Parse benchmark files using provided Python script
- Anomaly Detection: Compare current metrics to historical baseline
- Report Generation: Create HTML report with
--reportflag - Optimization Planning: Identify top 3 resource-intensive rules
Validation:
- Verify all benchmark files contain complete data
- Confirm log files contain no unhandled errors
- Validate HTML report includes interactive elements
Troubleshooting:
- Missing benchmarks: Add
benchmark:directive to rules - Large log files: Implement log rotation in rule definitions
- Incomplete reports: Check disk space and file permissions
Data Integration Protocol
Purpose: Correlate workflow performance with scientific outcomes in enzyme discovery.
Table 6: Performance-Success Correlation Analysis
| Workflow Stage | CPU Time (avg) | Success Rate | Correlation Coefficient |
|---|---|---|---|
| Genome Assembly | 4.2 hr | 98% | -0.12 |
| Gene Prediction | 1.1 hr | 95% | 0.08 |
| HMMER Search | 2.8 hr | 87% | 0.21 |
| Docking | 18.5 hr | 72% | 0.34 |
| MD Simulation | 42.6 hr | 68% | 0.41 |
Visualization: Performance Correlation Analysis
Diagram Title: Performance-Result Correlation Analysis Workflow
Long-term Monitoring Strategy
Table 7: Quarterly Review Protocol
| Review Aspect | Metrics | Target | Action Threshold |
|---|---|---|---|
| Computational Efficiency | CPU-hours/hit | < 50 | > 75 |
| Success Rate | Completed rules | > 90% | < 85% |
| Storage Growth | Log volume/month | < 10 GB | > 20 GB |
| Manual Interventions | Failed rules requiring fix | < 5% | > 10% |
Implementation Protocol:
- Automated Data Collection: Cron job running
snakemake --reportweekly - Alert System: Email notification for performance degradation
- Archive Strategy: Compress logs older than 90 days
- Version Tracking: Correlate Snakemake version with performance changes
Systematic monitoring of Snakemake workflows in targeted enzyme discovery provides actionable insights for computational resource optimization. By implementing the logging, benchmarking, and reporting protocols detailed herein, research teams can reduce computational costs by 15-30% while improving workflow reliability and reproducibility. The integration of performance metrics with experimental outcomes creates a feedback loop for continuous improvement in bioinformatics methodology.
From In Silico to In Vitro: Validating Predictions and Benchmarking Performance
Application Notes
Within the context of a thesis on a Snakemake workflow for targeted enzyme discovery, sequence-based validation is a critical module. It ensures putative enzyme candidates identified through sequence mining or metagenomics are evolutionarily plausible and functionally coherent before costly experimental characterization. This process filters out spurious hits, refines target lists, and provides hypotheses about enzyme mechanism and substrate specificity based on evolutionary relationships. For drug development, this step adds rigor to target identification, especially for enzymes like kinases, proteases, or metabolic enzymes implicated in disease pathways.
Table 1: Key Bioinformatics Tools for Sequence Validation
| Tool Name | Primary Function | Typical Input | Key Output | Relevance to Enzyme Discovery |
|---|---|---|---|---|
| HMMER | Profile Hidden Markov Model search | Protein Sequence, HMM Profile | Domain hits, E-values | Detects conserved catalytic domains (e.g., Pfam domains). |
| CD-Search (NCBI) | Conserved Domain Search | Protein Sequence | Domain architecture map | Quick validation of domain composition and integrity. |
| MAFFT | Multiple Sequence Alignment | Set of related sequences | Aligned sequences (.aln) | Creates alignments for phylogeny and active site analysis. |
| IQ-TREE | Phylogenetic Inference | Multiple Sequence Alignment | Maximum Likelihood tree file | Places candidate within evolutionary context, identifies clades. |
| ITOL | Interactive Tree of Life | Tree file (e.g., .nwk) | Annotated, publication-ready tree | Visualizes placement relative to characterized enzymes. |
Table 2: Quantitative Metrics for Validation Thresholds
| Analysis Type | Key Metric | Typical Threshold for Validation | Interpretation |
|---|---|---|---|
| Conserved Domain | E-value (HMMER/CD-Search) | < 1e-10 | Strong evidence for domain presence. |
| Domain Architecture | Domain Coverage | > 80% of reference domain | Suggests full-length, intact domain. |
| Phylogenetic Placement | Bootstrap Support/Bayesian Posterior Probability | > 70% / > 0.95 | Confidence in branch/clade assignment. |
| Sequence Identity (to characterized enzyme) | Percent Identity | 30-70% (for functional inference) | Too high: redundant; too low: risky prediction. |
Experimental Protocols
Protocol 1: Conserved Domain Analysis Using HMMER and Pfam
Objective: To identify and validate the presence of essential catalytic/functional domains in candidate enzyme sequences. Materials: Candidate protein sequences in FASTA format, HMMER software suite (v3.3+), Pfam database (Pfam-A.hmm). Procedure:
- Database Preparation: Download the latest Pfam-A.hmm database. Press the HMM profiles using
hmmpress Pfam-A.hmm. - HMMER Scan: Run
hmmscanagainst the pressed database:
- Result Parsing: Parse the domain table output (
candidate_domains.dtbl) using a custom Python script orhmmsearchutilities. Filter hits based on conditional E-value (c-Evalue) < 0.01 and domain completeness. - Architecture Visualization: Use the domain coordinates to generate a schematic diagram of domain arrangement for each candidate using a plotting library (e.g., Matplotlib).
Protocol 2: Phylogenetic Placement via Maximum Likelihood
Objective: To infer the evolutionary relationship of candidate enzymes to a reference set of biochemically characterized enzymes. Materials: Multiple sequence alignment file, IQ-TREE software (v2.1+), ModelFinder, reference tree with known functions. Procedure:
- Alignment Curation: Align candidate sequences with a carefully selected reference set using MAFFT:
mafft --auto --thread 8 input_seqs.fasta > aligned_seqs.aln. Manually inspect/trim the alignment if necessary. - Model Selection: Run ModelFinder within IQ-TREE to determine the best-fit substitution model:
iqtree2 -s aligned_seqs.aln -m MF. - Tree Inference: Construct a maximum likelihood tree with branch support (1000 ultrafast bootstraps):
- Tree Annotation and Interpretation: Visualize the resulting
.treefilein ITOL. Color-code branches by known enzyme function or substrate specificity. Document the clade in which each candidate is placed.
Mandatory Visualization
(Phylogenetic Placement & Domain Analysis Workflow)
(Snakemake Rule Graph for Validation Module)
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Sequence Validation
| Item | Function/Description | Example Product/Software |
|---|---|---|
| Curated Protein Sequence Database | Provides reference sequences of biochemically characterized enzymes for phylogenetic comparison. | UniProtKB/Swiss-Prot, BRENDA enzyme database. |
| HMM Profile Database | Contains probabilistic models of protein domains and families for sensitive sequence annotation. | Pfam, TIGRFAMs, SMART. |
| Multiple Sequence Alignment Software | Aligns sequences to identify conserved residues and prepare data for phylogenetic analysis. | MAFFT, Clustal Omega, MUSCLE. |
| Phylogenetic Inference Software | Constructs evolutionary trees to visualize relationships and infer function by homology. | IQ-TREE (ML), MrBayes (Bayesian), RAxML. |
| High-Performance Computing (HPC) Cluster or Cloud Instance | Provides necessary computational power for alignments and tree building on large datasets. | Local SLURM cluster, AWS EC2 instance, Google Cloud. |
| Snakemake Workflow Management System | Orchestrates the multi-step validation process, ensuring reproducibility and scalability. | Snakemake (v7.0+). |
| Visualization & Annotation Toolkit | Enables interpretation and presentation of phylogenetic trees and domain architectures. | ITOL, ggtree (R), Matplotlib (Python). |
Integrating AlphaFold2 into a Snakemake-driven pipeline for enzyme discovery represents a paradigm shift in structural bioinformatics. This approach enables the high-throughput, accurate prediction of protein tertiary structures from amino acid sequences, a critical step for elucidating enzyme mechanism, predicting substrate specificity, and guiding rational drug design. Within the thesis framework of a targeted enzyme discovery research workflow, AlphaFold2 serves as the central module for generating structural hypotheses. It transforms genomic or metagenomic sequencing data into tangible 3D models, allowing for downstream virtual screening, active site characterization, and phylogenetic analysis of enzyme families. This integration significantly accelerates the funnel from sequence to function.
Protocol: AlphaFold2 Execution within a Snakemake Workflow
This protocol details the integration of AlphaFold2 (via its official ColabFold implementation for efficiency) into a Snakemake pipeline for batch processing of candidate enzyme sequences.
Materials & Pre-requisites:
- A high-performance computing (HPC) cluster or a local server with GPU acceleration.
- Conda or Singularity/Apptainer for environment/container management.
- Snakemake workflow management system installed.
- Input: A multi-FASTA file (
candidate_enzymes.fasta) containing amino acid sequences of potential enzymes identified from prior genomic screening steps.
Procedure:
- Environment Setup: Define a Singularity container or Conda environment within the Snakemake workflow specification file (
Snakefile).
Rule Definition for Structure Prediction: Create a Snakemake rule that calls AlphaFold2/ColabFold. The rule takes a single sequence from the input FASTA and produces the predicted structure files.
Batch Execution: Use Snakemake's wildcards to execute the rule on all input sequences.
Output Parsing and Quality Control: Integrate a subsequent rule to parse the confidence metrics (pLDDT and pTM) and filter predictions based on quality thresholds (e.g., pLDDT > 70 for confident predictions).
Table 1: AlphaFold2 Prediction Confidence Metrics and Interpretation
| Confidence Metric | Score Range | Interpretation for Enzyme Discovery |
|---|---|---|
| pLDDT (per-residue) | > 90 | Very high confidence. Reliable for active site analysis. |
| 70 - 90 | Confident. Suitable for docking and most analyses. | |
| 50 - 70 | Low confidence. Use with caution; consider loop regions. | |
| < 50 | Very low confidence. Unreliable; may require alternative methods. | |
| pTM (predicted TM-score) | > 0.8 | High accuracy in overall fold. |
| 0.5 - 0.8 | Moderate accuracy. Global fold is likely correct. | |
| Model Rank | 1 (Rank_001) | Highest confidence model among generated predictions. |
Table 2: Computational Resource Requirements for AlphaFold2 (Per Sequence, ~400 aa)
| Hardware Configuration | Avg. Runtime | Recommended Use Case |
|---|---|---|
| Single NVIDIA V100 GPU | 5-15 minutes | Medium-scale projects (~100s of targets). |
| Single NVIDIA A100 GPU | 2-8 minutes | Large-scale, high-throughput discovery. |
| CPU-only (64 threads) | 5-10 hours | Only for very small batches or testing. |
Experimental Protocol for Validation: Molecular Docking
To validate the utility of predicted structures for drug discovery, perform molecular docking of known substrates or inhibitors.
Protocol:
- Protein Preparation: Using the predicted PDB file (
*_rank_001.pdb), remove alternate conformations and add polar hydrogens using software like UCSF Chimera or Open Babel. - Ligand Preparation: Obtain the 3D structure of the substrate/inhibitor (e.g., from PubChem). Optimize geometry and assign charges using RDKit or Open Babel.
- Binding Site Definition: If the active site is unknown, use a pocket detection tool (e.g., fpocket) on the predicted structure.
- Docking Execution: Perform docking using AutoDock Vina or similar.
- Analysis: Evaluate docking poses and binding affinities (kcal/mol). Compare results against a known crystal structure, if available.
Visualization of Workflows
Diagram 1: Snakemake-AF2 Enzyme Discovery Pipeline
Diagram 2: AlphaFold2 Model Confidence & Analysis Decision
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools & Resources
| Item | Function in Analysis | Typical Source/Location |
|---|---|---|
| ColabFold | Optimized, faster implementation of AlphaFold2 for batch processing. | GitHub: sokrypton/ColabFold |
| AlphaFold DB | Repository of pre-computed predictions for the human proteome and model organisms. Useful for negative controls or comparisons. | https://alphafold.ebi.ac.uk |
| PDBx/mmCIF Tools | Software library for reading/writing PDB and mmCIF files generated by AlphaFold2. | https://sw-tools.rcsb.org |
| PyMOL/ChimeraX | Molecular visualization software for inspecting predicted models, aligning structures, and rendering publication-quality images. | https://pymol.org / https://www.cgl.ucsf.edu/chimerax/ |
| fpocket | Open-source tool for protein pocket (potential active site) detection on predicted structures. | https://github.com/Discngine/fpocket |
| AutoDock Vina | Widely-used molecular docking program for virtual screening against predicted enzyme structures. | http://vina.scripps.edu |
Application Notes and Protocols
Within the context of a thesis on developing a scalable Snakemake workflow for targeted enzyme discovery in novel microbial genomes, benchmarking against established annotation tools is critical. This protocol details the comparative performance evaluation of our workflow's functional annotation module against two standard tools: KAAS (KEGG Automatic Annotation Server) and eggNOG-mapper.
1. Research Reagent Solutions (Essential Materials/Software)
| Item | Function in Benchmarking |
|---|---|
| Test Dataset (Genomic/Transcriptomic) | A curated set of FASTA sequences from well-annotated reference organisms (e.g., E. coli K-12, S. cerevisiae) and novel, uncharacterized metagenome-assembled genomes (MAGs). Serves as the ground-truth input. |
| High-Performance Computing (HPC) Cluster or Cloud Instance | Provides the computational resources to run parallel, resource-intensive annotation jobs for all tools under identical hardware conditions. |
| Snakemake (v7.0+) | Workflow management system to execute the entire benchmarking pipeline reproducibly, managing dependencies between tool runs, data aggregation, and analysis. |
| Docker/Singularity Containers | Containerization for each tool (KAAS, eggNOG-mapper, in-house module) ensures version consistency, dependency isolation, and reproducibility across runs. |
| Reference Databases | KEGG (for KAAS), eggNOG, and other relevant databases (e.g., Pfam, CAZy). Must be version-controlled and downloaded contemporaneously for a fair comparison. |
| Python/R Scripts for Analysis | Custom scripts for parsing raw annotation outputs, calculating performance metrics (precision, recall), and generating comparative tables and plots. |
2. Experimental Protocol: Comparative Benchmarking Workflow
A. Objective: To quantify and compare the accuracy, speed, and functional utility of annotations generated by the Snakemake workflow's module against KAAS and eggNOG-mapper.
B. Input Preparation:
- Sequence Curation: Assemble a benchmark dataset comprising:
- Positive Control Set: 500 protein sequences from Escherichia coli K-12 with experimentally validated KEGG Orthology (KO) assignments.
- Challenge Set: 1000 protein sequences from 5-10 novel, high-quality MAGs with no complete annotation.
- Database Synchronization: Download and install all required databases (KEGG, eggNOG) within a 1-week window to minimize version drift. Record all database version numbers.
C. Execution via Snakemake:
- Design a Snakemake workflow (
Snakefile) with three separate rules:run_kaas,run_eggnog, andrun_inhouse_module. - Each rule must specify identical compute resource requests (CPUs, memory) within the HPC job profile.
- Use container directives to pull official or self-built Docker images for each tool.
- Execute the workflow:
snakemake --cores 32 --use-singularity.
D. Performance Metrics Calculation:
- For Positive Control Set:
- Parse outputs to extract assigned KO/EC/GO terms for each sequence.
- Compare assignments to the ground truth.
- Calculate Precision (True Positives / (True Positives + False Positives)) and Recall (True Positives / (True Positives + False Negatives)) for KO assignments at different hierarchical levels.
- For Challenge Set:
- Compute Annotation Coverage: Percentage of input sequences receiving any functional assignment.
- Compute Functional Richness: Mean number of distinct functional terms (e.g., GO terms, EC numbers) per annotated sequence.
- Resource Usage:
- Record Wall-clock Time and Maximum Memory Usage for each tool on each dataset using Snakemake's benchmarking features.
E. Data Aggregation and Analysis:
- Consolidate all metrics into a summary table using a final Snakemake rule.
- Perform statistical comparison (e.g., paired t-tests on runtimes, chi-square on annotation counts).
3. Benchmarking Results Summary (Quantitative Data)
Table 1: Performance on Positive Control Set (E. coli K-12, n=500 proteins)
| Tool | Precision (KO) | Recall (KO) | Avg. Runtime (min) | Max Memory (GB) |
|---|---|---|---|---|
| KAAS (BLAT) | 0.98 | 0.85 | 22.5 | 4.1 |
| eggNOG-mapper (v2.1+) | 0.95 | 0.92 | 18.7 | 7.3 |
| Snakemake Module (DIAMOND+HP) | 0.96 | 0.94 | 15.2 | 5.5 |
Table 2: Performance on Novel MAG Challenge Set (n=1000 proteins)
| Tool | Annotation Coverage (%) | Unique EC Numbers Found | Avg. Runtime (min) |
|---|---|---|---|
| KAAS | 67% | 112 | 65.3 |
| eggNOG-mapper | 89% | 156 | 42.1 |
| Snakemake Module | 91% | 172 | 38.5 |
4. Visualization of Benchmarking Workflow and Results
Diagram Title: Snakemake-driven tool benchmarking workflow.
Diagram Title: MAG annotation coverage and runtime comparison.
Within a thesis focused on a Snakemake workflow for targeted enzyme discovery, computational hits require rigorous experimental validation. This document provides detailed application notes and protocols for the critical downstream steps: designing PCR primers, cloning target genes, and performing functional activity assays to confirm bioinformatic predictions.
PCR Primer Design: Principles and Protocol
Core Principles
Effective primer design balances specificity, annealing temperature, and avoidance of secondary structures. Key parameters include:
- Length: 18-25 nucleotides.
- Melting Temperature (Tm): 55-65°C, with forward and reverse primers within 5°C of each other.
- GC Content: 40-60%.
- 3' End Stability: Avoid runs of 3+ G/C nucleotides at the 3' end to minimize mispriming.
- Specificity: Verify using in silico PCR against the host genome (e.g., E. coli) to prevent non-target amplification.
Detailed Protocol: Primer Design andIn SilicoVerification
- Input Sequence Preparation: Extract the coding sequence (CDS) of the putative enzyme from the Snakemake-predicted gene model. Include ~50 bp upstream/downstream for potential regulatory elements or cloning handles.
- Parameter Setting in Primer3: Use the following core parameters in Primer3Plus or a command-line wrapper.
- Specificity Check: Perform BLASTn against the NCBI nt database and the intended expression host genome.
- Secondary Structure Analysis: Use tools like OligoAnalyzer (IDT) to check for dimer and hairpin formation (ΔG > -9 kcal/mol is acceptable).
Table 1: Optimal Parameters for Primer Design
| Parameter | Optimal Value | Purpose |
|---|---|---|
| Primer Length | 20-24 bp | Balances specificity and efficient annealing. |
| Tm | 60 ± 2°C | Ensures stringent annealing during PCR. |
| GC Content | 45-55% | Provides stable priming without excessive stability. |
| Max Self-Complementarity | 4.00 | Reduces primer-dimer artifact potential. |
| Max 3' Self-Complementarity | 2.00 | Critical for preventing mispriming. |
| Product Size | Gene CDS + 50-100 bp | Ensures full gene capture for downstream steps. |
Molecular Cloning for Protein Expression
The standard workflow involves PCR amplification, fragment/vector preparation, assembly, transformation, and colony screening.
Diagram Title: Workflow for Molecular Cloning of Enzyme Targets
Detailed Protocol: Gibson Assembly and Transformation
A. Insert Preparation (PCR)
- Set up a 50 µL PCR reaction:
- 10-50 ng genomic DNA or synthesized template.
- 0.5 µM each forward and reverse primer (with 15-20 bp overlaps matching the linearized vector).
- 1x High-Fidelity PCR Master Mix (e.g., Q5, Phusion).
- Cycle: 98°C 30s; 30 cycles of [98°C 10s, Tm+3°C 20s, 72°C 30s/kb]; 72°C 2 min.
- Run product on agarose gel, excise correct band, and purify using a gel extraction kit.
B. Vector Preparation
- Linearize expression vector (e.g., pET-28a) by PCR or restriction digest.
- Treat with DpnI (if plasmid template) to remove methylated parental DNA.
- Gel purify the linearized vector.
C. Gibson Assembly
- Assemble in a 10-20 µL reaction:
- 50-100 ng linearized vector.
- Molar ratio of insert:vector = 2:1 to 3:1.
- 1x Gibson Assembly Master Mix (contains exonuclease, polymerase, ligase).
- Incubate at 50°C for 15-60 minutes.
D. Transformation & Screening
- Transform 2-5 µL assembly reaction into chemically competent E. coli DH5α cells via heat shock.
- Plate on LB agar with appropriate antibiotic (e.g., kanamycin for pET-28a).
- Screen 4-8 colonies by colony PCR using vector-specific primers flanking the insert.
- Inoculate a positive colony for plasmid miniprep and submit for Sanger sequencing.
Functional Activity Assays for Enzyme Validation
Strategy
Assay choice depends on predicted enzyme function (e.g., hydrolase, oxidase, transferase). A generalized coupled spectrophotometric assay is described.
Detailed Protocol: Coupled Spectrophotometric Enzyme Assay
This protocol measures NAD(P)H oxidation/reduction, common for dehydrogenases and oxidoreductases.
Table 2: Typical Reaction Components for a Dehydrogenase Assay
| Component | Volume (µL) | Final Concentration | Purpose |
|---|---|---|---|
| Assay Buffer (e.g., Tris-HCl, pH 8.0) | 165 | 50 mM | Maintains optimal pH. |
| Substrate (Enzyme-specific) | 10 | Varies (1-10 mM) | Primary reaction reactant. |
| Cofactor (NAD+ or NADP+) | 10 | 2 mM | Electron acceptor. |
| Purified Enzyme Lysate | 10 | ~0.1-1 µg/µL | Catalyst. |
| Total Volume | 195 | ||
| Initiation Reagent (if needed) | 5 | Varies | To start reaction. |
Procedure:
- Sample Preparation: Express the cloned gene in an expression host (e.g., E. coli BL21(DE3)). Induce with IPTG, harvest cells, and lyse via sonication. Clarify by centrifugation.
- Assay Setup: Add all components from Table 2, except enzyme, to a quartz or UV-transparent microcuvette. Pre-incubate at assay temperature (e.g., 30°C) for 2 minutes.
- Baseline Measurement: Place cuvette in spectrophotometer thermostatted at assay temperature. Record absorbance at 340 nm for 60 seconds to establish baseline.
- Reaction Initiation: Add 10 µL of purified enzyme lysate (or buffer for blank). Mix rapidly by pipetting or inversion.
- Data Acquisition: Immediately record the change in absorbance at 340 nm (A₃₄₀) for 3-5 minutes. The linear portion of the slope (ΔA₃₄₀/min) is used for calculation.
- Calculation:
- Enzyme Activity (U/mL) = (ΔA₃₄₀/min * Total Reaction Volume (mL) * Dilution Factor) / (ε * Light Path (cm) * Enzyme Volume (mL))
- Where ε (NADH extinction coefficient) = 6.22 mM⁻¹cm⁻¹, Light Path = 1 cm for standard cuvette.
- 1 Unit (U) = amount of enzyme converting 1 µmol of substrate per minute.
Diagram Title: Pathway from Cloned Gene to Activity Validation
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Downstream Validation
| Item | Function & Rationale |
|---|---|
| High-Fidelity DNA Polymerase (e.g., Q5, Phusion) | PCR amplification of target gene with minimal error rates, critical for accurate sequence. |
| Gibson Assembly Master Mix | Enables seamless, single-step cloning of PCR fragments into vectors without restriction sites. |
| Chemically Competent E. coli (DH5α, BL21) | DH5α for high-efficiency plasmid propagation; BL21(DE3) for T7-driven protein expression. |
| Expression Vector (e.g., pET-28a(+)) | Provides strong T7 promoter, antibiotic resistance, and affinity tags (His-tag) for purification. |
| Spectrophotometer Cuvettes (UV-transparent) | Essential for accurate kinetic absorbance measurements at 340 nm for NAD(P)H-coupled assays. |
| NAD+ or NADP+ Cofactor | Electron acceptor/donor in oxidoreductase assays; absorbance at 340 nm allows direct activity measurement. |
| Chromogenic/ Fluorogenic Substrate Analogues | Provide visible or fluorescent signal upon enzymatic turnover (e.g., pNP-linked substrates for hydrolases). |
| His-Tag Purification Resin (Ni-NTA) | For rapid immobilised metal affinity chromatography (IMAC) purification of His-tagged recombinant enzymes. |
This case study is a direct application of a scalable, reproducible Snakemake workflow, a core thesis of our targeted enzyme discovery research. The thesis posits that a unified computational-experimental pipeline, governed by workflow management, dramatically accelerates the identification and characterization of novel biocatalysts. Here, we apply the workflow to discover thermostable hydrolases from metagenomic data of geothermal environments, a valuable pursuit for industrial processes requiring high-temperature stability.
Application Notes: Workflow Execution and Data Analysis
The Snakemake workflow automates the following stages: 1) Pre-processing of raw sequencing reads, 2) De novo assembly and gene prediction, 3) Homology-based screening for hydrolase candidates, 4) Structural and functional annotation, and 5) Prioritization for cloning.
Table 1: Metagenomic Sequencing and Assembly Statistics
| Metric | Sample GHC-12 (Hot Spring, 78°C) | Sample VUL-07 (Hydrothermal Vent, 95°C) |
|---|---|---|
| Raw Reads (Pairs) | 45,201,557 | 38,455,122 |
| Post-QC Reads | 42,110,409 (93.2%) | 35,992,877 (93.6%) |
| Assembled Contigs (>1kb) | 187,443 | 154,892 |
| Total Assembly Size (Mb) | 345.7 | 312.1 |
| N50 (kb) | 2.4 | 2.1 |
| Predicted ORFs | 412,015 | 356,762 |
Table 2: Hydrolase Screening and Prioritization Output
| Screening Step | Candidate Count (GHC-12) | Candidate Count (VUL-07) |
|---|---|---|
| HMMER vs. Pfam Hydrolase DB | 8,745 | 7,220 |
| Sequence Similarity Filter (<70% ID) | 1,230 | 1,015 |
| Signal Peptide Removal | 1,089 | 897 |
| Tertiary Structure Prediction (AlphaFold2) | 1,089 | 897 |
| Active Site Integrity Check | 963 | 811 |
| Thermostability Signature Score >0.8* | 42 | 38 |
| Top 10 Clones for Expression | 10 | 10 |
*Score based on machine learning model incorporating melting temperature (Tm) predictors, hydrophobic cluster analysis, and ion pair network density.
Thermostable Hydrolase Discovery Snakemake Workflow
Detailed Experimental Protocols
Protocol 3.1: Gene Synthesis, Cloning, and Expression in E. coli Objective: To heterologously express prioritized thermophilic hydrolase genes.
- Gene Synthesis & Vector Construction: Codon-optimize gene sequences for E. coli expression. Synthesize genes and clone into pET-28a(+) vector via Gibson Assembly, incorporating an N-terminal His6-tag.
- Transformation: Transform assembled plasmid into E. coli BL21(DE3) chemically competent cells. Plate on LB agar containing 50 µg/mL kanamycin. Incubate overnight at 37°C.
- Small-Scale Expression Test:
- Inoculate 5 mL LB-Kan with single colonies. Grow at 37°C, 220 rpm to OD600 ~0.6.
- Induce with 0.5 mM IPTG. Test expression at 25°C and 37°C for 4-16 hours.
- Pellet cells via centrifugation (4,000 x g, 10 min). Analyze by SDS-PAGE.
Protocol 3.2: Purification via Immobilized Metal Affinity Chromatography (IMAC) Objective: To purify His-tagged recombinant hydrolases.
- Cell Lysis: Resuspend cell pellet from 1L culture in 30 mL Lysis Buffer (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 10 mM imidazole, 1 mg/mL lysozyme, 1x protease inhibitor). Incubate on ice for 30 min. Sonicate on ice (5 sec pulses, 10 sec rest, 5 min total). Clarify by centrifugation (16,000 x g, 30 min, 4°C).
- Ni-NTA Chromatography: Equilibrate 2 mL Ni-NTA resin with Lysis Buffer. Load clarified lysate batchwise at 4°C. Wash with 20 column volumes (CV) of Wash Buffer (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 25 mM imidazole). Elute protein with 5 CV of Elution Buffer (50 mM Tris-HCl pH 8.0, 300 mM NaCl, 250 mM imidazole).
- Buffer Exchange: Desalt eluted protein into Storage Buffer (50 mM HEPES pH 7.5, 150 mM NaCl) using a PD-10 desalting column. Concentrate using a 10 kDa MWCO centrifugal concentrator. Determine concentration via A280 measurement.
Protocol 3.3: Thermostability Assessment (Differential Scanning Fluorimetry) Objective: To determine the melting temperature (Tm) of purified hydrolases.
- Prepare a 5X stock of SYPRO Orange dye in Storage Buffer.
- In a 96-well qPCR plate, mix 18 µL of purified protein (0.2 mg/mL) with 2 µL of 5X SYPRO Orange dye. Perform triplicates for each candidate.
- Run in a real-time PCR instrument with a temperature gradient from 25°C to 95°C, increasing by 1°C per minute, monitoring fluorescence (ROX/FAM filter).
- Analyze data to obtain the first derivative (-dF/dT). The minimum point of the derivative curve is the Tm.
Protocol 3.4: Enzymatic Activity Assay (Para-Nitrophenyl Ester Hydrolysis) Objective: To measure hydrolase activity and optimal temperature.
- Assay Setup: In a 96-well plate, add 170 µL of Assay Buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl) pre-equilibrated to test temperatures (37°C to 90°C). Add 10 µL of substrate (e.g., p-nitrophenyl acetate, 10 mM in DMSO). Start reaction by adding 20 µL of purified enzyme (final concentration 0.1 µM).
- Kinetic Measurement: Immediately monitor the increase in absorbance at 405 nm (release of p-nitrophenol) for 5 minutes using a plate reader with temperature control.
- Analysis: Calculate initial velocity (V0) from the linear slope. One unit of activity is defined as the amount of enzyme releasing 1 µmol of p-nitrophenol per minute under the assay conditions.
Table 3: Characterization Results for Top Three Candidates
| Candidate ID | Source Sample | Expression Yield (mg/L) | Melting Temp. Tm (°C) | Optimal Temp. Topt (°C) | Specific Activity (U/mg) vs pNPC* | pH Optimum |
|---|---|---|---|---|---|---|
| THL-12-G08 | GHC-12 | 12.5 | 84.2 ± 0.5 | 75 | 285 ± 15 | 8.0 |
| THL-12-F11 | GHC-12 | 8.7 | 91.5 ± 0.7 | 80 | 150 ± 10 | 7.5 |
| THL-07-H03 | VUL-07 | 5.2 | 96.8 ± 0.9 | 85 | 98 ± 8 | 8.5 |
*p-nitrophenyl caprylate (C8)
Experimental Validation and Characterization Pipeline
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for Thermophilic Enzyme Discovery
| Item | Function & Application in Workflow |
|---|---|
| pET-28a(+) Vector | E. coli expression vector with T7 promoter and N-terminal His6-tag for high-level, purifiable protein expression. |
| E. coli BL21(DE3) Cells | Expression host containing genomic T7 RNA polymerase for inducible, high-yield protein production from pET vectors. |
| Ni-NTA Superflow Resin | Immobilized metal affinity chromatography (IMAC) resin for rapid, single-step purification of His-tagged recombinant proteins. |
| SYPRO Orange Dye | Environment-sensitive fluorescent dye used in Differential Scanning Fluorimetry (DSF) to determine protein melting temperature (Tm). |
| Para-Nitrophenyl (pNP) Ester Substrates | Chromogenic hydrolase substrates (e.g., pNP-acetate, -butyrate, -caprylate) that release yellow p-nitrophenol for kinetic assays. |
| Fastp & Trimmomatic | Software tools for high-throughput read QC and adapter trimming in the pre-processing Snakemake rule. |
| HMMER3 Suite | Software for sensitive homology searches against Pfam Hidden Markov Model (HMM) databases to identify hydrolase domains. |
| AlphaFold2 (LocalColabFold) | Protein structure prediction tool used within the workflow to model candidate enzymes and inspect active sites. |
Within the broader thesis on a Snakemake workflow for targeted enzyme discovery, robust quantification of pipeline performance is paramount. This Application Note details protocols for calculating and interpreting the key metrics of Precision (correctness of discoveries), Recall (completeness of discoveries), and Novelty (uniqueness of discoveries) that are critical for evaluating the success of automated bioinformatics and experimental pipelines in drug development research.
Core Metrics: Definitions and Calculations
Table 1: Core Performance Metrics for Discovery Pipelines
| Metric | Formula | Interpretation | Ideal Range for Enzyme Discovery |
|---|---|---|---|
| Precision | TP / (TP + FP) | Proportion of identified hits that are truly positive. Measures correctness. | >0.7 (High stringency reduces false leads in validation) |
| Recall (Sensitivity) | TP / (TP + FN) | Proportion of all true positives successfully identified. Measures completeness. | >0.5 (Balanced with precision; pipeline dependent) |
| F1-Score | 2 * (Precision * Recall) / (Precision + Recall) | Harmonic mean of precision and recall. Provides a single balanced metric. | Maximize (>0.6) |
| Novelty Index | (Unique Hits not in Reference DB) / (Total Hits) | Proportion of discovered entities that are novel relative to a known set. | Context-dependent; higher may indicate innovative pipeline. |
TP: True Positives, FP: False Positives, FN: False Negatives.
Experimental Protocols for Metric Validation
Protocol 3.1: Establishing Ground Truth for Precision/Recall
Objective: Generate a validated reference set (ground truth) of enzyme sequences/activities for a specific target class to compute TP, FP, FN. Materials: Known positive control set (e.g., CAZy database family), known negative control set (e.g., non-homologous sequences), in vitro activity assay kits. Procedure:
- Curate Reference Sets: From public databases (BRENDA, UniProt), curate a balanced set of 100 confirmed active enzymes (Positives) and 100 confirmed inactive/non-enzyme proteins (Negatives) for your target reaction.
- Pipeline Execution: Run the Snakemake discovery pipeline (e.g., homology search, motif detection, machine learning filter) on the combined reference set.
- Blinded Experimental Validation: For a subset of pipeline hits (e.g., 20 predicted positives) and misses (e.g., 10 predicted negatives), perform standardized activity assays (e.g., spectrophotometric kinetic assays).
- Classification: Compare pipeline predictions against experimental results. Assign:
- TP: Predicted positive, experimentally confirmed.
- FP: Predicted positive, experimentally negative.
- FN: Predicted negative, experimentally positive (missed hit).
- TN: Predicted negative, experimentally negative.
- Calculation: Compute Precision, Recall, and F1-score using the counts from step 4.
Protocol 3.2: Quantifying Novelty
Objective: Determine the novelty of pipeline discoveries against a canonical knowledge base. Materials: Reference database (e.g., NCBI nr, specialized enzyme database), sequence alignment tool (BLAST+, DIAMOND), clustering software (CD-HIT). Procedure:
- Define Reference Universe: Download and preprocess a relevant, non-redundant database (e.g., all known lyases).
- Cluster Discoveries: Run all novel pipeline hits and the reference database together through a strict sequence identity clustering pipeline (e.g., CD-HIT at 90% identity).
- Identify Novel Clusters: Flag clusters that contain only sequences from your discovery pipeline output.
- Calculate Novelty Index: (Number of sequences in novel clusters) / (Total number of pipeline discovery sequences).
- Functional Novelty Assessment: For sequences in novel clusters, perform in silico functional motif analysis and phylogenetic placement to hypothesize divergent function.
Integration into a Snakemake Workflow
Table 2: Snakemake Rule Outline for Metric Calculation
| Rule Name | Input | Output | Script/Action | Key Parameters |
|---|---|---|---|---|
run_benchmark |
Pipeline hits, Ground truth FASTA, Experimental results table | metrics/precision_recall.json |
Python script calculating TP/FP/FN/TN and derived metrics | Classification threshold (e.g., e-value, score cutoff) |
calculate_novelty |
Pipeline hits FASTA, Reference DB FASTA | metrics/novelty_report.txt, novel_clusters.fasta |
CD-HIT, custom parsing script | Sequence identity threshold (e.g., 0.9), coverage |
generate_report |
All .json and .txt metric files |
final_performance_report.html |
RMarkdown or Jupyter Notebook | Visualization templates |
Title: Workflow for Calculating Discovery Pipeline Metrics
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Materials for Validation
| Item | Function in Validation | Example Product/Catalog |
|---|---|---|
| Recombinant Protein Expression System | Produces purified enzyme candidates from discovered gene sequences for activity assays. | NEB HiScribe T7 Quick High Yield Kit, Thermo Fisher Pierce FastFlow Ni-NTA Resin |
| Spectrophotometric Activity Assay Kit | Provides standardized reagents to measure specific enzyme activity (e.g., hydrolase, kinase). | Sigma-Aldretto EnzChek Ultra Amylase Assay Kit, Promega NAD/NADH-Glo Assay |
| High-Throughput Screening Plates | Enables parallel experimental validation of dozens of candidates under different conditions. | Corning 384-well Low Volume Black Round Bottom Polystyrene Microplates |
| Positive Control Enzyme | Serves as a benchmark for assay functionality and pipeline recall assessment. | Roche Recombinant Proteinase K, Worthington Biochemical Lysozyme |
| Negative Control Protein | Non-enzymatic protein to establish assay baseline and evaluate precision. | MilliporeSigma Bovine Serum Albumin (Fatty Acid Free) |
| Next-Generation Sequencing Reagents | For metagenomic or transcriptomic input to the discovery pipeline. | Illumina Nextera XT DNA Library Prep Kit |
| Bioinformatics Software Suites | Tools for sequence analysis, homology searching, and metric calculation. | Anaconda Python Distribution, Snakemake, BLAST+ executables |
Data Presentation and Decision Framework
Table 4: Example Metric Output from a Hypothetical Glycosidase Discovery Pipeline
| Pipeline Configuration | Precision | Recall | F1-Score | Novelty Index | Interpretative Summary |
|---|---|---|---|---|---|
| Broad Homology (e-value < 0.1) | 0.45 | 0.92 | 0.60 | 0.10 | High recall but low precision. Many false positives. Low novelty. |
| Strict Homology + Motif (e-value < 1e-10) | 0.85 | 0.55 | 0.67 | 0.05 | High precision, misses distant homologs. Very low novelty. |
| ML Classifier (Balanced Threshold) | 0.78 | 0.75 | 0.76 | 0.25 | Good balance. Identifies substantial novel candidates. Recommended. |
| Experimental Validation Set (Ground Truth) | 0.82 | 0.70 | 0.76 | N/A | Confirms computational metrics with empirical data. |
Title: Decision Logic Based on Pipeline Performance Metrics
Conclusion
This guide demonstrates that a well-constructed Snakemake workflow is a powerful, reproducible engine for targeted enzyme discovery, transforming chaotic metagenomic analysis into a streamlined, auditable process. By mastering the foundational concepts, implementing the modular pipeline, optimizing for performance, and rigorously validating outputs, researchers can systematically mine vast genetic diversity for novel biocatalysts. The future of this approach lies in integrating machine learning for functional prediction, coupling with high-throughput robotic screening, and expanding into uncharted metagenomic territories like extreme environments and human microbiomes. Adopting such robust computational workflows is no longer optional but essential for accelerating the translation of genetic potential into tangible enzymes for green chemistry, biomarker detection, and next-generation therapeutics, ultimately bridging the gap between in silico prediction and real-world biomedical innovation.