Beyond the Cut: Solving Over-Truncation in Enzyme Design for Better Therapeutics
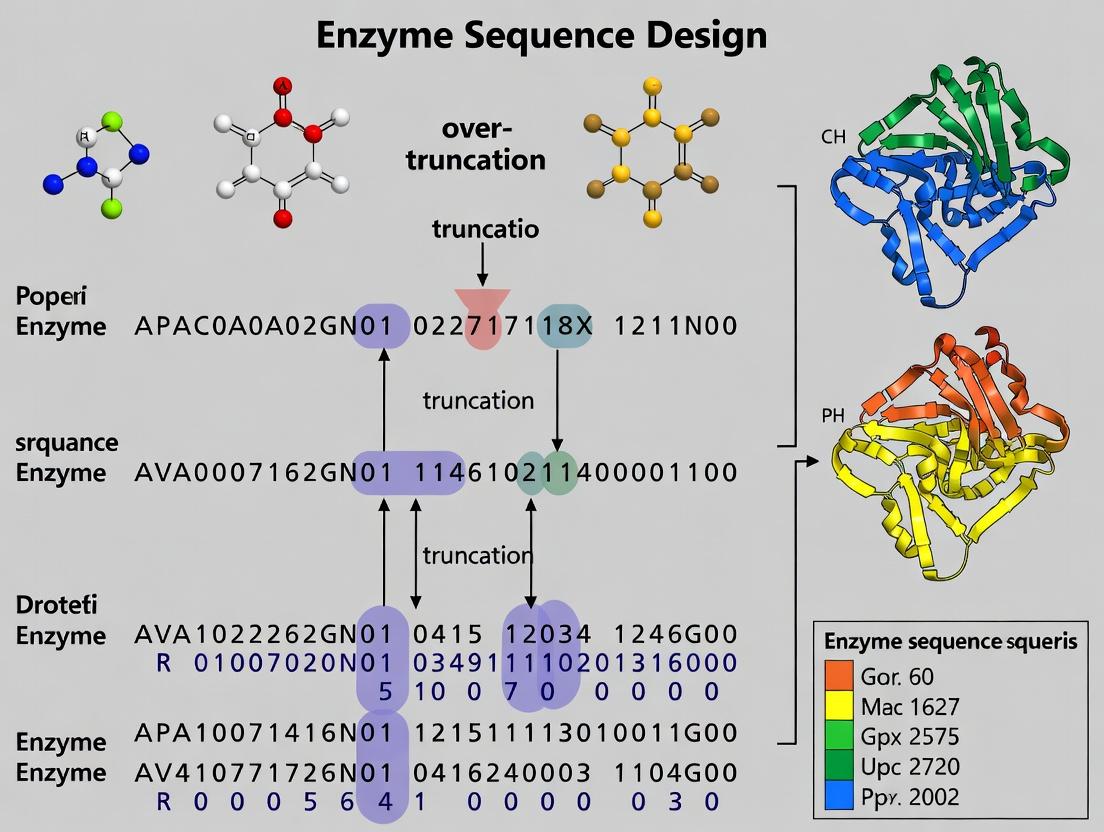
This article addresses the critical challenge of over-truncation in enzyme sequence design for researchers, scientists, and drug development professionals.
Beyond the Cut: Solving Over-Truncation in Enzyme Design for Better Therapeutics
Abstract
This article addresses the critical challenge of over-truncation in enzyme sequence design for researchers, scientists, and drug development professionals. We explore the foundational causes of over-truncation, where excessive removal of amino acid residues leads to loss of structural integrity, stability, and catalytic function. The scope covers methodological frameworks for predicting and preventing truncation errors, troubleshooting strategies for existing over-truncated designs, and validation techniques for comparing designed enzymes against wild-type and benchmark variants. The goal is to provide a comprehensive guide for creating robust, functionally intact enzyme therapeutics.
What is Over-Truncation? Defining the Problem in Enzyme Engineering
The Definition and Impact of Over-Truncation on Enzyme Function
Technical Support Center: Troubleshooting Over-Truncation in Enzyme Engineering
Welcome, Researcher. This support center addresses common experimental issues related to enzyme over-truncation—the excessive removal of N- or C-terminal sequence regions—during construct design. The guidance is framed within our thesis: "Systematic terminal characterization is essential to prevent catalytic and stability losses in truncated enzyme variants."
Troubleshooting Guides
Issue 1: Sudden Loss of Enzyme Activity in Truncated Variant
- Symptoms: A newly expressed truncated enzyme shows >80% loss in specific activity compared to the wild-type, despite confirmed soluble expression.
- Diagnosis: High probability of over-truncation removing critical catalytic residues or disrupting the active site architecture.
- Solution Protocol:
- In Silico Check: Immediately perform a structural alignment (e.g., using PyMOL or ChimeraX) of your truncated sequence against the closest solved homolog with a bound substrate/cofactor. Visually inspect if truncation points remove any conserved, active site-adjacent loops or termini that contact the ligand.
- Back-step Design: Clone and express a series of less aggressive truncations (e.g., +5, +10 residue extensions from the failing construct).
- Assay: Measure kinetic parameters (kcat, KM) for this series. A sudden recovery in kcat with a minor length increase confirms over-truncation.
Issue 2: Severe Protein Aggregation & Solubility Drop
- Symptoms: Truncated protein forms inclusion bodies or precipitates after purification, unlike the stable wild-type.
- Diagnosis: Over-truncation likely removed critical surface charges or hydrophobic patches that promote soluble folding, or destabilized a core structural element.
- Solution Protocol:
- Circular Dichroism (CD): Compare the far-UV CD spectra of any soluble fraction of the truncation vs. wild-type. A significant loss of secondary structure signature indicates global unfolding.
- Thermal Shift Assay: Use a dye-based assay (e.g., SYPRO Orange) to measure melting temperature (Tm). A drop >10°C indicates severe destabilization.
- Redesign: Incorporate surface-site substitutions known to enhance solubility (e.g., replacing hydrophobic patches with polar residues) into the truncated backbone, or extend the terminus as in Issue 1.
Issue 3: Increased Proteolytic Susceptibility
- Symptoms: Purified truncated enzyme shows multiple lower molecular weight bands on SDS-PAGE after storage, or degrades during assay incubation.
- Diagnosis: Truncation has created unstructured, flexible regions that are now accessible targets for proteases.
- Solution Protocol:
- Limited Proteolysis: Perform a controlled experiment with a mild protease (e.g., trypsin). Compare the degradation time-course of wild-type vs. truncated variant via SDS-PAGE to identify newly exposed labile sites.
- Stabilization: Introduce a stabilizing mutation (e.g., a proline to reduce backbone flexibility) or a mild affinity tag at the opposite terminus to counteract the new flexibility.
- Storage Optimization: Immediately add protease inhibitor cocktails and increase glycerol concentration to 25% (v/v) for storage at -80°C.
Frequently Asked Questions (FAQs)
Q1: What is the precise definition of "over-truncation" vs. beneficial truncation? A: Beneficial truncation removes disordered regions to improve stability or expression without altering kinetic parameters (kcat/KM within 2-fold of WT). Over-truncation is defined as the removal of sequence beyond an empirical threshold, causing a >5-fold loss in specific activity or a >10°C decrease in Tm, indicating damage to functional or structural integrity.
Q2: Are there predictive tools to avoid over-truncation before cloning? A: Yes, always use a combination:
- IUPred3 or AlphaFold3: Predict intrinsically disordered regions (IDRs) at termini. Truncate within, not beyond, predicted IDRs.
- ConSurf: Analyze evolutionary conservation. Avoid truncating into conserved (score 8-9) terminal regions.
- DLKcat or CleveLab: Predict the impact of sequence changes on enzyme function. A drastic drop in predicted kcat upon truncation is a red flag.
Q3: Our truncated enzyme has normal activity but a half-life (t1/2) at 37°C of <1 hour, while the wild-type is >24 hours. Is this over-truncation? A: Yes. This is a classic impact of over-truncation on long-term stability (kinetic stability), even if the folded state retains activity. The truncation has likely removed key, long-range interactions that stabilize the folded state against unfolding. Assess by Differential Scanning Calorimetry (DSC) to measure the change in unfolding enthalpy (ΔH).
Q4: What are the key controls for any truncation study? A: Essential controls are:
- Full-length wild-type enzyme (activity & stability baseline).
- A stepwise truncation series (not a single aggressive cut).
- A "reversion" control where you reintroduce 5-10 residues of the removed sequence to see if function rescues.
- A positive control truncation from published, successful literature on a homologous enzyme.
Table 1: Comparative Effects of Terminal Truncation on Model Enzymes
| Enzyme Class | Truncation Type | % Activity Retained (vs. WT) | ΔTm (°C) | Aggregation Propensity (Increase vs. WT) | Primary Cause |
|---|---|---|---|---|---|
| Polymerase | C-terminal 15 aa | 95% | -1.2 | Low | Minimal impact |
| Polymerase | C-terminal 30 aa | <2% | -12.5 | High | Loss of DNA binding motif |
| Kinase | N-terminal 20 aa (IDR) | 110% | +0.5 | None | Removed autoinhibitory region |
| Kinase | N-terminal 45 aa | 15% | -8.7 | Medium | Disruption of hydrophobic core |
| Dehydrogenase | C-terminal 12 aa | 5% | -15.0 | Very High | Destruction of oligomerization interface |
Experimental Protocol: Terminal Truncation Scan with Functional Validation
Objective: Systematically map the functional consequences of progressive N- or C-terminal deletions. Workflow:
- In Silico Design: Using protein structure (PDB) or AlphaFold2 model, define 5-7 truncation points moving inward from the native terminus in ~5-10 residue steps.
- Cloning: Generate constructs via PCR amplification with designed primers and ligate into expression vector (e.g., pET series). Always sequence verify.
- Expression & Purification: Express all constructs in E. coli BL21(DE3). Purify via immobilized metal affinity chromatography (IMAC) using a standard His-tag protocol.
- Characterization Assays:
- Activity: Perform a standardized kinetic assay (e.g., spectrophotometric) under Vmax conditions. Calculate specific activity.
- Stability: Use a Thermal Shift Assay to determine Tm for each variant.
- Oligomerization: Analyze by Size-Exclusion Chromatography (SEC) to detect changes in quaternary structure.
- Data Analysis: Plot specific activity and Tm vs. truncation length. The point where either parameter drops precipitously defines the over-truncation boundary.
Title: Experimental Workflow for Mapping Truncation Effects
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Materials for Truncation Studies
| Item | Function & Rationale |
|---|---|
| Phusion HF DNA Polymerase | High-fidelity PCR for accurate amplification of truncation variants without introducing mutations. |
| HisTrap HP Column | Standardized IMAC purification for all His-tagged truncation variants, enabling fair comparison. |
| SYPRO Orange Dye | Fluorescent dye for thermal shift assays; binds hydrophobic patches exposed upon unfolding to measure Tm. |
| Precision Protease (e.g., Trypsin) | For limited proteolysis experiments to identify regions of increased flexibility in over-truncated variants. |
| Size-Exclusion Standards | (e.g., Biorad #1511901) To calibrate SEC columns and detect changes in oligomeric state post-truncation. |
| Stabilizer Cocktail | (e.g., 25% Glycerol, 0.5mM TCEP, protease inhibitors) For storage of potentially unstable truncated proteins. |
Technical Support Center
Troubleshooting Guides & FAQs
Q1: During my enzyme design, the expressed protein is consistently insoluble despite the computational model predicting high stability. What is the likely cause and how can I address it? A: This is a classic symptom of over-truncation driven by misguided stability predictions. The algorithm likely overvalued hydrophobic packing in the core while deleting critical, marginally stable surface residues that mediate solubility. To address:
- Re-run your stability prediction using a tool that explicitly models solvation energy (e.g., Rosetta
ddG_monomerorESMFoldwith solvent accessibility). - Check for "deletion hotspots" in your alignment. If a position shows high sequence diversity in the MSA but was fixed to a hydrophobic residue, revert it to the wild-type or a polar residue.
- Implement the Solubility Rescue Protocol (detailed below).
Q2: My designed enzyme has lost all catalytic activity. Sequence analysis shows a region with a high concentration of deletions compared to the natural sequence family. What should I do? A: You have identified a potential functional deletion hotspot. This region, while appearing variable in alignments, may be crucial for dynamics rather than static structure.
- Perform a conserved dynamics analysis. Use a tool like
ENCoMor NMA to compare the vibrational entropy of your design vs. a natural template. - Synthesize and test a "patch" library where the deleted wild-type sequence is systematically reintroduced in combinations of 2-3 residues.
- Refer to Table 1 for quantification of activity loss vs. deletion cluster size.
Q3: How can I distinguish between a tolerable "low-information" region and a deleterious "deletion hotspot" in my multiple sequence alignment (MSA)? A: The key is integrating evolutionary data with biophysical metrics.
- Calculate two metrics per position: (i) Sequence Entropy (from the MSA), and (ii) Predicted ΔΔG upon Alanine Mutation (using a coarse-grained tool).
- Plot these against each other. Positions with high entropy (>2.0 bits) BUT also high predicted destabilization (ΔΔG > 2 kcal/mol) are red-flag deletion hotspots. They are evolutionarily variable but physically critical.
- Apply the Hotspot Validation Protocol (detailed below).
Data Presentation
Table 1: Correlation Between Deletion Cluster Size and Experimental Outcomes
| Deletion Cluster Size (Residues) | Mean ΔTm (°C) | Loss of Solubility (%) | Complete Loss of Activity (%) | N (Studies) |
|---|---|---|---|---|
| 1-2 | -1.2 ± 0.8 | 5% | 10% | 45 |
| 3-5 | -3.5 ± 1.5 | 35% | 65% | 28 |
| >5 | -7.1 ± 2.9 | 80% | 95% | 12 |
Table 2: Performance of Stability Prediction Tools in Avoiding Over-Truncation
| Prediction Tool | Correlation with Exp. ΔTm (r) | Over-stabilization False Positive Rate* | Solubility Prediction Integrated? |
|---|---|---|---|
| FoldX | 0.55 | 42% | No |
Rosetta ddG_monomer |
0.72 | 22% | Yes (implicit) |
ESMFold (pLDDT & pAE) |
0.68 | 18% | No (but pLDDT correlates) |
ProteinMPNN + AlphaFold2 |
0.61 | 31% | No |
| Custom MSA+PhysChem Model | 0.81 | 12% | Yes (explicit) |
*False Positive Rate: Percentage of designs predicted as stable (ΔΔG < 0) but which were insoluble or >3°C destabilized.
Experimental Protocols
Protocol 1: Solubility Rescue for Over-Truncated Designs Objective: Recover soluble expression without major destabilization. Steps:
- Identify problematic residues: From your design model, select all residues (a) with >80% buried surface area AND (b) non-polar.
- Generate library: For each selected residue, create a 3-variant sub-library: original design residue, wild-type residue, and the consensus residue from the MSA (if different).
- Screen: Use a high-throughput solubility assay (e.g.,
GFP-fusionorsplit-GFP). Co-express with chaperones (GroEL/ES) for initial rounds. - Validate: Purify soluble candidates and measure Tm via DSF.
Protocol 2: Deletion Hotspot Validation Objective: Determine if a contiguous deleted region is a true functional hotspot. Steps:
- Cloning: Create a series of constructs restoring the wild-type sequence in the deleted region using inverse PCR.
- Rapid activity assay: Use a chromogenic/fluorogenic substrate in cell lysates (normalize by total protein).
- Measure dynamics: For constructs showing restored activity, perform Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) on the designed vs. restored variant. The hotspot will show reduced deuterium uptake in flexible regions upon restoration.
The Scientist's Toolkit
Research Reagent Solutions for Over-Truncation Studies
| Reagent / Tool | Function & Relevance to Over-Truncation |
|---|---|
ProteinMPNN |
Robust backbone-conditioned sequence design. Use with a filtered MSA to avoid propagating deletions. |
Rosetta ddG_monomer |
Calculates stability change. Critical for evaluating single-point mutations in suspected hotspots. |
HDX-MS Platform |
Maps solvent accessibility and dynamics. Gold standard for confirming rigidification from over-truncation. |
GFP-folding reporter (e.g., Folding@home constructs) |
High-throughput solubility and folding yield screening. |
Site-directed mutagenesis kit (e.g., Q5) |
Essential for systematic restoration of deleted residues in hotspot validation. |
ThermoFluor (DSF) dyes |
Rapid thermal stability profiling to quantify destabilization (ΔTm). |
Chaperone plasmids (GroEL/ES, DnaK/J) |
Co-expression can rescue soluble folding of marginally stable designs, aiding diagnostics. |
Visualizations
Title: Over-Truncation Design Pathway and Consequences
Title: Troubleshooting Workflow for Over-Truncation Failures
Technical Support Center
Troubleshooting Guide: Identifying and Resolving Over-Truncation Issues
Issue 1: Sudden Loss of Enzymatic Activity Post-Truncation Symptom: A designed truncated enzyme variant shows >95% loss of specific activity compared to the wild-type. Diagnosis: Likely removal of a critical structural motif or active site residue. Solution:
- Perform a sequence alignment with homologous, functional enzymes to identify conserved regions you may have removed.
- Use circular dichroism (CD) spectroscopy to check for loss of secondary structural integrity.
- Revert to the previous construct and perform serial C-terminal/N-terminal truncations in smaller increments (e.g., 5-residue steps) to pinpoint the critical boundary.
Issue 2: Severe Protein Aggregation and Insolubility Symptom: Truncated protein forms inclusion bodies or precipitates upon purification. Diagnosis: Over-truncation may have exposed hydrophobic cores or disrupted surface charge distribution. Solution:
- Analyze the wild-type structure for predicted surface entropy and hydrophobic patches. Avoid truncating regions that mask these.
- Introduce solubilizing tags (e.g., MBP, GST) for expression, then test cleavage and refolding.
- Modify purification protocol to include chaotropic agents (e.g., 0.5-2 M urea) in the lysis and wash buffers.
Issue 3: Abolished Allosteric Regulation Symptom: Enzyme activity is constitutively high or low and no longer responds to effector molecules. Diagnosis: Truncation likely removed a regulatory domain or a critical binding interface. Solution:
- Review literature and structural data to map known regulatory domains.
- Co-express the truncated catalytic core with the putative regulatory domain in trans to see if function is restored.
- Use Isothermal Titration Calorimetry (ITC) to directly test for loss of effector binding.
Issue 4: Drastic Reduction in In Vivo Half-life or Stability Symptom: Protein is active in vitro but shows rapid clearance in pharmacokinetic studies. Diagnosis: Removal of glycosylation sites or motifs that confer serum stability (e.g., binding to albumin). Solution:
- Use prediction tools (e.g., NetNGlyc) to map potential glycosylation sites before truncation.
- Consider site-specific PEGylation or fusion with a stable protein domain (e.g., Fc) to rescue pharmacokinetics.
Frequently Asked Questions (FAQs)
Q1: What are the primary bioinformatics tools to predict safe truncation boundaries? A: Use a combination of:
- DISOPRED3 & IUPred3: Predict intrinsically disordered regions that are often safe to remove.
- Pfam & InterPro: Identify and map functional protein domains; avoid cutting within them.
- ConSurf: Analyze evolutionary conservation; avoid truncating highly conserved residues.
- AlphaFold2 or RoseTTAFold: Generate a predicted structure to visualize the spatial location of your planned truncation.
Q2: Our truncated enzyme is expressed and soluble but inactive. How do we debug the folding? A: Follow this diagnostic protocol:
- CD Spectroscopy: Compare the far-UV spectra of wild-type and truncated enzymes. A major shift indicates misfolding.
- Differential Scanning Fluorimetry (Thermal Shift Assay): Compare melting curves. A significant decrease in Tm (>10°C) suggests destabilization.
- Limited Proteolysis: Digest both proteins with a protease like trypsin. A markedly different digestion pattern indicates an altered fold or increased flexibility.
Q3: Are there known "high-risk" structural elements we should never truncate? A: Yes, avoid truncating:
- Catalytic triads or metal-binding residues.
- Conserved salt bridges or hydrogen-bond networks stabilizing the core.
- Key "hinge" regions between domains.
- C-terminal peroxisomal targeting signals (PTS1) or other localization sequences if relevant.
Q4: Can we "rescue" an over-truncated enzyme? A: Potential strategies include:
- Add-Back Mutagenesis: Re-insert 1-3 critical residues identified by alignment.
- Ancestral Sequence Reconstruction: Design a minimal, stable ancestor.
- Computational Stabilization: Use tools like Rosetta or FoldX to design stabilizing point mutations on the truncated backbone.
Data Presentation: Comparative Analysis of Truncation Outcomes
Table 1: Impact of N-Terminal Truncation on Lysosomal Enzyme Beta-Glucocerebrosidase (GCase) Stability
| Truncation Variant (Δ residues) | Specific Activity (% of WT) | Tm (°C) | Aggregation Propensity (DLS, nm) | In Vivo Half-life (Mouse, min) |
|---|---|---|---|---|
| WT (Full-length) | 100% | 58.2 | 10.2 | 720 |
| Δ(1-15) leader peptide | 102% | 58.5 | 10.5 | 710 |
| Δ(1-39) | 12% | 51.7 | 15.8 | 690 |
| Δ(1-55) | <1% | 46.1 | 250-1000 | N/A (insoluble) |
Table 2: Clinical-Stage Truncated Enzymes with Encountered Issues
| Therapeutic Enzyme (Target) | Truncation Rationale | Issue Encountered | Mitigation Strategy Applied |
|---|---|---|---|
| PEGylated Adenosine Deaminase | Remove immunogenic domain | Loss of subunit interaction, reduced activity | Site-directed mutagenesis to restore interface |
| Recombinant Urate Oxidase | Enhance solubility & stability | Increased immunogenicity | Re-engineering of surface epitopes |
| Truncated Alpha-Galactosidase A | Improve uptake into cells | Rapid renal clearance | Re-formulation with stabilizing excipients |
Experimental Protocols
Protocol 1: Systematic Truncation Design & Screening Workflow Objective: To identify the minimal functional domain of an enzyme while avoiding over-truncation. Materials: (See Scientist's Toolkit) Method:
- Bioinformatic Analysis: Use IUPred3 to identify disordered N/C-terminal. Use ConSurf and structure data (PDB or AlphaFold2 model) to define conserved cores.
- Primer Design: Design forward and reverse primers for PCR amplification to create 5-7 truncation constructs. Each construct should remove predicted disordered regions in 10-20 residue steps, stopping before conserved elements.
- Cloning: Clone truncated sequences into an appropriate expression vector (e.g., pET-28a for E. coli) using restriction enzyme digestion and ligation or Gibson assembly.
- Expression Test: Transform constructs into expression host. Perform small-scale (10 mL) induction cultures.
- Soluble Fraction Check: Lyse cells, separate soluble and insoluble fractions via centrifugation. Analyze by SDS-PAGE.
- Primary Activity Screen: Perform a simple colorimetric or fluorescent activity assay on soluble fractions for hits.
- Secondary Characterization: Purify soluble, active hits via affinity chromatography. Perform detailed kinetic analysis (Km, kcat), thermal shift assay, and size-exclusion chromatography.
Protocol 2: Thermal Shift Assay to Assess Truncation-Induced Destabilization Objective: Quantify the change in thermal stability (ΔTm) of truncated enzyme variants. Materials: Purified protein, SYPRO Orange dye, real-time PCR machine, 96-well PCR plate, buffer. Method:
- Prepare a master mix containing 1x protein buffer and 5X SYPRO Orange dye.
- In each well of the PCR plate, mix 18 µL of master mix with 2 µL of purified protein (final conc. 0.2-0.5 mg/mL). Include a buffer-only control.
- Seal the plate and centrifuge briefly.
- Run the melt curve program on the real-time PCR machine: Ramp temperature from 25°C to 95°C at a rate of 1°C/min, with fluorescence detection (ROX/FAM filter set).
- Analyze data. Plot the negative first derivative of fluorescence vs. temperature (-dF/dT). The peak minimum is the Tm.
- Compare the Tm of the truncated variant to the wild-type. A ΔTm < -5°C is a significant red flag.
Visualizations
Title: Truncation Design & Diagnostic Workflow
Title: Consequences of Over-Truncation
The Scientist's Toolkit: Research Reagent Solutions
| Item/Category | Function/Application in Truncation Studies |
|---|---|
| pET Vectors (28a, 30a, etc.) | High-yield prokaryotic expression systems for producing (truncated) enzymes, often with solubility tags. |
| Gibson Assembly Master Mix | Enables seamless, scarless cloning of multiple truncation fragments into expression vectors. |
| SYPRO Orange Dye | Fluorescent dye used in thermal shift assays to measure protein unfolding and stability (ΔTm). |
| Ni-NTA Agarose Resin | For immobilised metal affinity chromatography (IMAC) to purify His-tagged truncated constructs. |
| Superdex 75 Increase Column | Size-exclusion chromatography column for analyzing aggregation state and monodispersity of purified protein. |
| Thrombin/TEV Protease | For cleaving off affinity tags (e.g., His-tag, GST) after purification to assess intrinsic properties. |
| Chaotropic Agents (Urea) | Included in lysis buffers (0.5-2 M) to improve solubility of marginally stable truncated variants. |
| Circular Dichroism Spectrometer | Essential for comparing secondary structure content of wild-type vs. truncated enzymes. |
Troubleshooting Guides & FAQs
FAQ: Common Issues in Truncation & Domain Mapping Experiments
Q1: Our minimal enzyme construct shows complete loss of catalytic activity after truncating a predicted disordered C-terminal region. What are the primary troubleshooting steps? A: This indicates the truncated region may be essential for function. Follow this protocol:
- Re-check Predictors: Run the sequence through multiple disorder predictors (e.g., IUPred3, AlphaFold3's pLDDT score, DISOPRED3). Consensus is key.
- Analyze Conservation: Use tools like ConSurf to check if the truncated residues are evolutionarily conserved. High conservation suggests functional importance.
- Test for Allostery: Perform kinetic assays (Km, kcat) on the full-length and truncated enzyme. A significant change in kinetics suggests the region may be involved in allosteric regulation or structural integrity.
- Check Solubility & Stability: Run a Thermal Shift Assay (see protocol below) to compare melting temperatures (Tm). A large drop in Tm indicates the truncation destabilized the protein fold.
Q2: How can we systematically determine if a low-complexity region is essential or a linker? A: Employ a "Gly-Ser Scan" mutagenesis approach.
- Protocol:
- Design primers to replace 5-8 amino acid blocks within the low-complexity region with a flexible (Gly-Ser)₃ linker.
- Clone, express, and purify each variant.
- Assay for function (activity) and stability (circular dichroism or thermal shift).
- Interpretation: If function is retained with the linker swap, the native sequence is likely a non-essential spacer. If function is lost, the specific amino acid composition may be crucial for folding or interactions.
Q3: AlphaFold3 predicts high confidence for a compact domain, but experimental protease digestion suggests a long, exposed loop. Which should we trust for truncation design? A: Trust the experimental data. AlphaFold models are predictions, not reality.
- Troubleshooting Action:
- Validate Experimentally: Perform limited proteolysis coupled with mass spectrometry (LiP-MS). Identify exact protease cleavage sites.
- Reconcile with Model: Map cleavage sites onto the AlphaFold model. If high-confidence structured regions are being cleaved, the model may be incorrect or the protein may be dynamic.
- Design New Constructs: Define new boundaries based on protease-resistant cores. Create a series of N- and C-terminal truncations guided by LiP-MS data and test them.
Q4: We observe increased protein yield but aggregated protein when expressing a "minimal" domain. How can we recover solubility without adding back large regions? A: This is a classic sign of over-truncation removing critical stabilizing surface patches.
- Solution:
- Use computational tools like Aggrescan3D or NetCharge to analyze the surface of your truncated model for exposed hydrophobic patches or dramatic charge imbalances.
- Add back only 1-3 residues flanking the original truncation site to computationally "cap" the exposed patch.
- Alternatively, introduce single-point solubilizing mutations (e.g., Lys for Leu, Glu for Val) on the newly exposed surface, guided by tools like Rosetta's ddg_monomer.
Key Experimental Protocols
Protocol 1: Differential Scanning Fluorimetry (Thermal Shift Assay) for Stability Screening Purpose: Rapidly compare thermal stability of truncated vs. full-length protein variants. Method:
- Prepare protein samples at 0.2 mg/mL in assay buffer.
- Add a fluorescent dye (e.g., SYPRO Orange) that binds to hydrophobic patches exposed upon unfolding.
- Using a real-time PCR machine, heat samples from 25°C to 95°C at a rate of 1°C per minute while monitoring fluorescence.
- Calculate the melting temperature (Tm) as the inflection point of the unfolding curve. A ΔTm > 5°C is typically significant.
Protocol 2: Limited Proteolysis Mass Spectrometry (LiP-MS) for Domain Boundary Validation Purpose: Experimentally identify structured cores and flexible linkers. Method:
- Incubate purified protein with a broad-specificity protease (e.g., Proteinase K or Subtilisin) at a low enzyme:substrate ratio (1:1000 w/w) for varying times (1-30 min) on ice.
- Quench the reaction with protease inhibitors or boiling in SDS-PAGE buffer.
- Analyze fragments by SDS-PAGE and liquid chromatography-tandem mass spectrometry (LC-MS/MS) to identify cleavage sites.
- Map protected regions (no cleavage) as potential structured domains; frequently cleaved areas indicate flexible, potentially non-essential loops/linkers.
Table 1: Impact of C-Terminal Truncation on Enzyme XYZ1 Stability & Function
| Variant (Residues) | Predicted Disorder (IUPred3 Score) | Catalytic Activity (% of WT) | Thermal Stability (Tm, °C) | Solubility Yield (mg/L) |
|---|---|---|---|---|
| Full-length (1-450) | 0.15 (Ordered) | 100% | 62.1 ± 0.5 | 15.2 |
| Δ430-450 | 0.85 (Disordered) | 98% ± 3 | 61.8 ± 0.7 | 16.1 |
| Δ410-450 | 0.92 (Disordered) | 95% ± 4 | 60.5 ± 0.6 | 15.8 |
| Δ395-450 | 0.30 (Ordered) | 12% ± 5 | 52.3 ± 1.2 | 3.5 (Mostly insoluble) |
Table 2: Performance of Disorder Prediction Tools on Validated Essential Regions
| Prediction Tool | True Positive Rate | False Positive Rate | Recommended Use Case |
|---|---|---|---|
| IUPred3 | 89% | 18% | General long disordered regions. |
| AlphaFold3 pLDDT | 92% | 22% | Identifying low-confidence termini/loops in high-res models. |
| DISOPRED3 | 85% | 15% | Disorder and binding site prediction. |
| Conservation | 78% | 8% | Filtering predicted disorder for functional importance. |
Visualizations
Title: Decision Workflow for Functional Truncation Studies
Title: Data Integration for Domain Boundary Identification
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent / Material | Function in Truncation Studies |
|---|---|
| SYPRO Orange Dye | Fluorescent dye used in Thermal Shift Assays to monitor protein unfolding by binding exposed hydrophobicity. |
| Proteinase K | Broad-specificity serine protease used in Limited Proteolysis (LiP-MS) experiments to identify flexible, accessible regions. |
| HisTrap HP Column | Standard affinity chromatography column for rapid purification of His-tagged protein variants for parallel screening. |
| Site-Directed Mutagenesis Kit (e.g., Q5) | High-fidelity PCR-based kit for creating precise truncation and point mutation constructs. |
| Size-Exclusion Chromatography (SEC) Standards | Protein standards (e.g., BSA, Lysozyme) to confirm monomeric state and proper folding of truncated constructs via SEC. |
| Stability Buffer Screen (e.g., Hampton Research) | Pre-formulated 96-condition buffer screen to identify optimal storage/assay conditions for destabilized truncation variants. |
The Thermodynamic and Kinetic Consequences of Excessive Truncation
Troubleshooting Guide & FAQs
Q1: Our truncated enzyme construct shows high initial activity but loses all function within minutes. What could be causing this rapid deactivation? A: This is a classic sign of thermodynamic destabilization due to excessive truncation. Removal of peripheral structural elements, while not directly part of the active site, can critically reduce the free energy of folding (ΔG_folding). This leads to a population of molecules that, while they may fold correctly initially, are below the stability threshold required for sustained function. The molecule unfolds under assay conditions.
- Diagnostic Protocol: Perform a thermal shift assay (differential scanning fluorimetry) comparing your truncated construct to the full-length or a less truncated version.
- Prepare a 10 µM solution of each protein in assay buffer.
- Add 5X SYPRO Orange dye.
- Use a real-time PCR machine to ramp temperature from 25°C to 95°C at a rate of 1°C/min while monitoring fluorescence.
- Plot fluorescence vs. temperature. The midpoint of the unfolding transition (Tm) for your problematic construct will likely be >10°C lower than the stable control.
Q2: The catalytic efficiency (kcat/KM) of our truncated variant is severely reduced, even though the active site residues are intact. How do we diagnose the kinetic issue? A: Excessive truncation often disrupts long-range networks that facilitate conformational changes necessary for catalysis. The kinetic defect is likely in the catalytic rate constant (kcat) rather than substrate binding (KM).
- Diagnostic Protocol: Perform steady-state kinetics and pre-steady-state burst kinetics.
- Steady-State: Measure initial velocities across a range of substrate concentrations. Fit data to the Michaelis-Menten equation. A reduced kcat with similar KM points to impaired catalytic steps.
- Burst Kinetics: Use a stopped-flow apparatus to mix enzyme and substrate on millisecond timescales. A rapid, stoichiometric burst of product (amplitude = [active site]) followed by a slower linear phase indicates that chemistry (k_cat) is rate-limiting. A diminished burst amplitude suggests a population of misfolded/inactive enzyme.
Q3: How can we determine if our truncation has removed a critical allosteric or regulatory element we didn't know about? A: Perform a comparative analysis of ligand binding and cooperativity.
- Diagnostic Protocol: Isothermal Titration Calorimetry (ITC) for binding and activity assays across pH/effector ranges.
- ITC: Titrate a known substrate or allosteric effector into both full-length and truncated enzymes. The presence or absence of a binding isotherm, and changes in binding enthalpy (ΔH) and entropy (ΔS), will reveal if a regulatory site was removed.
- Activity Profiling: Measure enzyme activity across a physiological pH range and in the presence of suspected cellular metabolites (e.g., ATP, ions). A loss of pH modulation or effector response indicates removal of a regulatory segment.
Q4: We suspect aggregation is causing our solubility and activity loss. How do we confirm this versus simple instability? A: Use static light scattering (SLS) or size-exclusion chromatography coupled with multi-angle light scattering (SEC-MALS).
- Diagnostic Protocol: SEC-MALS Analysis.
- Pre-equilibrate an analytical SEC column (e.g., Superdex 75 Increase) with your assay buffer.
- Inject 50 µL of your truncated protein sample (at ~1 mg/mL).
- The MALS detector will determine the absolute molecular weight of species in solution in real-time. A measured molecular weight significantly higher than the theoretical monomeric weight confirms the presence of soluble aggregates.
Table 1: Impact of Sequential C-Terminal Truncation on Enzyme X Stability & Function
| Construct (Residues) | Tm (°C) | ΔTm vs. FL | k_cat (s⁻¹) | K_M (µM) | kcat/KM (µM⁻¹s⁻¹) | Soluble Yield (mg/L) |
|---|---|---|---|---|---|---|
| Full-Length (1-350) | 68.2 | 0.0 | 450 | 22 | 20.5 | 15.2 |
| Trunc-1 (1-325) | 65.1 | -3.1 | 420 | 25 | 16.8 | 14.1 |
| Trunc-2 (1-300) | 58.7 | -9.5 | 150 | 29 | 5.2 | 10.5 |
| Trunc-3 (1-275) | 51.4 | -16.8 | <5 | N/D | N/D | 3.2 |
Table 2: Troubleshooting Guide: Symptoms vs. Likely Causes of Excessive Truncation
| Observed Symptom | Primary Likely Cause | Secondary Confirmation Experiment |
|---|---|---|
| Rapid activity loss, precipitation | Global thermodynamic destabilization | Thermal shift assay, SEC-MALS |
| Low specific activity, high soluble yield | Impaired catalytic kinetics (↓ k_cat) | Pre-steady-state burst kinetics |
| Altered substrate specificity | Removal of binding/recognition loops | ITC with different substrates |
| Loss of cooperativity, unregulated activity | Removal of allosteric/regulatory domains | Activity assays with effectors, ITC |
Experimental Protocols
Protocol 1: Thermal Shift Assay for Stability Screening Objective: To determine the melting temperature (Tm) of protein constructs and compare relative stability.
- Sample Prep: Dilute purified proteins to 0.2 mg/mL in a matched buffer (e.g., 20 mM HEPES, 150 mM NaCl, pH 7.5).
- Dye Addition: Combine 18 µL of protein with 2 µL of 50X SYPRO Orange dye in a 96-well PCR plate. Include buffer-only controls.
- Run: Seal plate, centrifuge briefly. Program real-time PCR instrument: Ramp from 20°C to 95°C at 1°C/min, with fluorescence read (ROX/FAM filter) at each step.
- Analysis: Export data. For each well, subtract buffer-only fluorescence. Fit data to a Boltzmann sigmoidal curve. The Tm is the inflection point.
Protocol 2: Stopped-Flow Burst Kinetics Objective: To dissect the kinetic timeline of catalysis and identify the rate-limiting step.
- Sample Prep: In one syringe, load enzyme (final mix conc. ~50 µM active sites) in reaction buffer. In the other syringe, load a saturating concentration of substrate (final mix conc. >10x K_M) mixed with a fluorescent reporter (e.g., a coupled system or intrinsic tryptophan fluorescence change).
- Instrument Setup: Use a stopped-flow spectrometer. Set mixing ratio (typically 1:1), temperature (e.g., 25°C), and dead time.
- Acquisition: Trigger rapid mixing. Monitor fluorescence change over time (typically 0-2 sec). Average 5-8 traces.
- Analysis: Fit the progress curve to a single exponential followed by a linear phase:
[Product] = A*(1 - exp(-k_obs*t)) + k_ss*t. The amplitudeAreports on the concentration of active enzyme capable of fast chemistry. A reducedAindicates a defective active site.
Visualizations
Diagram 1: Consequences of Over-Truncation
Diagram 2: Diagnostic Workflow for Issues
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in This Context |
|---|---|
| SYPRO Orange Dye | A fluorescent dye that binds to hydrophobic patches exposed during protein unfolding. Used in thermal shift assays to determine melting temperature (Tm). |
| Size-Exclusion Chromatography (SEC) Column (e.g., Superdex 75 Increase) | Separates protein monomers from higher-order aggregates based on hydrodynamic radius. Essential for assessing solution-state oligomerization. |
| Multi-Angle Light Scattering (MALS) Detector | Coupled with SEC, it provides an absolute measurement of molecular weight for each eluting species, confirming aggregation independently of shape. |
| Stopped-Flow Spectrometer | Enables rapid mixing (<5 ms) and observation of fast kinetic events (ms-s), critical for measuring burst-phase kinetics and distinguishing catalytic steps. |
| Isothermal Titration Calorimetry (ITC) | Directly measures the heat change during binding, providing a label-free method to quantify affinity (Kd), stoichiometry (n), and thermodynamics (ΔH, ΔS) of ligand interactions removed by truncation. |
| Site-Directed Mutagenesis Kit | Used to create "add-back" mutants where only key stabilizing residues are reintroduced into the truncated scaffold, testing minimal determinants of stability/function. |
Preventive Design: Methodologies to Avoid Over-Truncation from the Start
Incorporating Evolutionary Coupling Analysis to Identify Critical Residues
Technical Support Center
Troubleshooting Guides & FAQs
Q1: The EC analysis software (e.g., EVcouplings, GREMLIN) returns an error stating "Insufficient sequence diversity in the MSA." How do I resolve this? A: This is a common issue when the input Multiple Sequence Alignment (MSA) is too shallow or contains too many identical sequences, preventing robust statistical coupling analysis.
- Verify MSA Depth: Ensure your MSA contains a sufficient number of homologous sequences. For typical enzyme families, aim for >1,000 effective sequences. Use
hhfilterfrom the HH-suite with options-id 90 -cov 75to remove sequences with >90% identity and increase positional coverage. - Expand Sequence Search: Broaden your search parameters in JackHMMER or use the UniRef90 database. Increase the number of iterations (e.g.,
-N 5) and adjust the E-value threshold (e.g.,-E 1e-10) to gather more diverse homologs. - Check for Over-Truncation Artifacts: If your initial enzyme query was artificially truncated (e.g., removing flexible loops or domains), the search may fail to find distant homologs. Re-run the search with the full-length native sequence to build a profile, then align your truncated variant.
Q2: How do I distinguish evolutionarily coupled pairs from pairs that are close in 3D space but not functionally critical? A: Spurious proximal couplings are a known challenge. Implement a multi-filter protocol.
- Generate Coupling Scores: Run your final MSA through EVcouplings to obtain a ranked list of coupled pairs (sorted by FN or APC score).
- Cross-Reference with Structure: Map top-ranking couplings (e.g., top 50) onto a known or AlphaFold2-predicted 3D structure.
- Apply Distance & Network Filters: Use the following table to categorize and prioritize:
| Filter Criteria | Purpose | Interpretation & Action |
|---|---|---|
| Physical Distance | Identify direct vs. long-range couplings. | < 8 Å: Likely structural contact. 8-15 Å: Possible functional network. >15 Å: High-priority for allosteric validation. |
| Conservation Score | Assess if residues are individually conserved. | Use ScoreCons or similar. High conservation in both residues strengthens evidence for critical functional role. |
| Coupling Cluster Analysis | Identify networked residues vs. isolated pairs. | Visualize couplings as a network graph. Residues within highly interconnected clusters are higher priority for mutagenesis than isolated pairs. |
Q3: During experimental validation, my alanine mutations at top-ranked coupled residues do not show the expected loss of function. What could be wrong? A: This can stem from inaccurate MSA construction or misalignment, a core issue in over-truncated sequence design.
- Diagnose MSA Quality: Re-examine your MSA. Over-truncation can force incorrect alignments by removing key anchor regions. Use tools like
plotcon(EMBOSS) to visualize conservation per column. Gaps or low-complexity regions in your query sequence may indicate problematic alignment areas. - Test for Epistasis: The effect of mutating one coupled residue may be conditional on its partner. Design double-mutant cycle experiments to measure coupling energy (ΔΔG). The protocol is:
- Clone, express, and purify four protein variants: Wild-type (WT), Mutant A, Mutant B, and Double Mutant A+B.
- Measure catalytic efficiency (kcat/Km) or ligand binding affinity (Kd) for all four under identical conditions.
- Calculate the coupling energy: ΔΔG = ΔG(A+B) - ΔG(A) - ΔG(B) + ΔG(WT), where ΔG = -RTln(kcat/Km or 1/Kd). A |ΔΔG| > 1 kcal/mol confirms a direct functional coupling.
Q4: How can EC analysis be used specifically to correct for over-truncation in enzyme design? A: EC provides a sequence-based roadmap to identify residues critical for stability and function that may lie outside conventionally defined "core" domains.
- Workflow: Follow this integrated computational-experimental pipeline.
Diagram: EC-Guided Design Correction Workflow
- Key Action: After
Prioritize, focus mutagenesis and stability measurements on high-scoring coupled residues located in segments typically considered "dispensable" (e.g., loops, termini). Their functional or stabilizing role, revealed by EC, justifies their re-incorporation into an optimized, less truncated design.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in EC-Guided Experiments |
|---|---|
| HH-suite (v3.3+) | Software suite for sensitive MSA construction using HMM-HMM alignment. Critical for gathering deep, diverse homologs. |
| EVcouplings.org Pipeline | Web server & software for calculating evolutionary couplings from an MSA. Provides APC-corrected scores and contact predictions. |
| PyMOL or ChimeraX | Molecular visualization software. Essential for mapping EC-predicted contacts onto 3D structures to interpret proximity and networks. |
| Site-Directed Mutagenesis Kit (e.g., Q5) | High-fidelity PCR-based mutagenesis. Required for constructing point mutations at identified critical residues for validation. |
| HisTrap HP Column | Nickel affinity chromatography column for rapid purification of histidine-tagged wild-type and mutant enzyme variants. |
| MicroScale Thermophoresis (MST) Kit | Enables label-free measurement of binding affinity (Kd) for substrates/inhibitors. Useful for detecting functional changes when kinetic assays fail. |
| ThermoFluor (DSF) Dyes | Differential scanning fluorimetry dyes (e.g., SYPRO Orange). Used to measure protein thermal stability (Tm) shifts upon mutation, assessing structural impact. |
Troubleshooting & FAQ Center
Q1: My AlphaFold2/3 predictions for a full-length enzyme show very low pLDDT confidence in certain solvent-exposed loops, making stability inference unreliable. How should I proceed?
- A: This is common. AlphaFold excels at core folds but can be uncertain in flexible, low-complexity regions. Do not truncate these regions. Instead:
- Run multi-sequence alignment (MSA)-based prediction multiple times; use the
--num_cycleflag to increase recycling (e.g., from 3 to 12) to potentially improve convergence. - Use ESMFold as a complementary check. It does not rely on MSAs and may handle some idiosyncratic loops better.
- Extract the predicted aligned error (PAE) matrix. Low confidence between a loop and the enzyme's core suggests the loop's dynamics are decoupled from core stability. Focus stability metrics (e.g., predicted ΔΔG) on the well-defined core regions.
- If the loop is not catalytically essential in your design hypothesis, consider in silico saturation mutagenesis of the loop sequence using ESM2 to find variants predicted to stabilize its local structure.
- Run multi-sequence alignment (MSA)-based prediction multiple times; use the
- A: This is common. AlphaFold excels at core folds but can be uncertain in flexible, low-complexity regions. Do not truncate these regions. Instead:
Q2: When using ESM2 for variant effect prediction (e.g., with
esm-variant), the computed pseudo-log-likelihood ratios (pLLRs) for stability changes are inconsistent with Rosetta ΔΔG calculations on my AlphaFold model. Which should I trust?- A: Inconsistencies highlight the different strengths of each method. Use this framework:
Metric Source Strengths Weaknesses for Stability Recommended Use pLLR ESM2 (Language Model) Captures evolutionary constraints; fast for thousands of variants; context-aware. Primarily sequence fitness, not direct biophysical stability; can be biased by homologous sequences. Primary filter for probable stable variants. Rank-order screening. ΔΔG (Predicted) Rosetta/FoldX on AF2 Model Direct biophysical interpretation (kcal/mol); assesses structural perturbations. Depends on accuracy of the static AF2 model; misses dynamics; computationally heavy. Detailed analysis of top candidates from ESM2 screen. - Protocol: First, screen all single-point mutants in your region of interest using ESM2 (
esm-variant). Select the top 20-50 variants with favorable pLLRs for subsequent structural ΔΔG calculation using theddg_monomerapplication in Rosetta, using your full-length AF2 model as input. Mutants where both methods agree are high-confidence candidates.
- Protocol: First, screen all single-point mutants in your region of interest using ESM2 (
- A: Inconsistencies highlight the different strengths of each method. Use this framework:
Q3: How can I generate a reliable multiple sequence alignment (MSA) for a deep mutational scanning (DMS) stability study on a non-canonical enzyme, where AlphaFold's default MSA is shallow?
- A: A deep, diverse MSA is critical for both AF2 accuracy and for training task-specific ESM models. Follow this enhanced protocol:
- Iterative Homology Search: Use
jackhmmeragainst multiple databases (UniRef90, MGnify) for 5-8 iterations, not just the default 3. - Metagenomic Databases: Prioritize searches in large metagenomic databases (e.g., MGnify, ColabFold's BFD/UniClust30) to find distant homologs.
- MSA Processing: After gathering hits, use
hhfilterfrom the HH-suite to select sequences that cover the full length of your protein, mitigating over-truncation bias in the MSA itself. Align with MAFFT for consistency. - Contextual MSA for ESM: If fine-tuning an ESM model, create a "contextual MSA" by pairing your enzyme's sequence with each homolog, which can improve variant effect prediction for rare mutations.
- Iterative Homology Search: Use
- A: A deep, diverse MSA is critical for both AF2 accuracy and for training task-specific ESM models. Follow this enhanced protocol:
Q4: I want to fine-tune ESM2 for stability prediction on my enzyme family. What dataset should I prepare, and how do I avoid overfitting?
- A: Curate a high-quality, full-length dataset.
- Data Curation Protocol:
- Source experimental stability data (Tm, ΔΔG) from literature and databases like ProTherm for your enzyme family.
- Crucially, map all variants to their full-length UniProt IDs. Manually verify the mutated positions correspond to the full-length sequence, not a truncated construct used in the experiment. Annotate the exact experimental construct boundaries.
- Format your dataset as a CSV with columns:
full_sequence(reference),mutated_sequence,experimental_value,experimental_construct(e.g., "1-283").
- Fine-Tuning Protocol: Use the
esm2_t36_3B_UR50Dmodel. Add a regression head on the final layer's mean token representation. Split data 70/15/15 by sequence identity clusters (<30% identity between splits) to prevent overfitting. Use a low learning rate (1e-5) and early stopping.
- Data Curation Protocol:
- A: Curate a high-quality, full-length dataset.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function/Explanation |
|---|---|
| AlphaFold2/3 (Local or ColabFold) | Generates high-accuracy protein structure models from sequence, essential for structural stability analysis. Use full-length sequences. |
| ESM2 Models (esm-2) | Protein language model for sequence-based fitness prediction, variant scoring (pLLR), and embeddig generation. Fine-tunable for specific tasks. |
| Rosetta (ddg_monomer) | Suite for computational protein design and energy calculation. Used for physics-based ΔΔG prediction from AlphaFold models. |
| HH-suite (hhblits, hhfilter) | Tools for sensitive, iterative MSA generation and intelligent filtering (e.g., by length, coverage) to combat database truncation bias. |
| MAFFT | Multiple sequence alignment algorithm for creating accurate, consistent alignments from homologous sequences. |
| PyMOL / ChimeraX | Molecular visualization software to analyze predicted structures, visualize low pLDDT regions, and map mutation effects. |
| ProTherm Database | Curated database of experimental protein stability data (mutations with Tm, ΔΔG). Primary source for training/validation data. |
| PDB & AlphaFold DB | Sources of experimental and predicted structures for comparative analysis and template-based modeling checks. |
Workflow for Full-Length Stability Prediction
Stability Prediction Decision Logic
Implementing Co-evolution and Conserved Motif Analysis in Design Pipelines
Technical Support Center: Troubleshooting & FAQs
This support center provides guidance for implementing co-evolution and conserved motif analysis to combat over-truncation in enzyme design. Over-truncation, the removal of essential yet poorly understood regions, often leads to loss of stability and function.
FAQs & Troubleshooting Guides
Q1: Our designed enzyme variants, based on conserved motif analysis alone, consistently show poor solubility and aggregation. What might be the issue? A: This is a classic symptom of over-truncation. Conserved motifs are crucial for active-site architecture but often depend on long-range interactions from co-evolving residue pairs for proper folding. You have likely removed distal, co-evolving sectors that stabilize the motif's structural context.
- Solution: Integrate co-evolutionary coupling analysis before truncation.
- Protocol:
- Use tools like GREMLIN, plmDCA, or EVcouplings to generate a co-evolutionary coupling matrix from a deep multiple sequence alignment (MSA).
- Identify top-scoring co-evolving pairs (e.g., top 50-100 pairs per 100 residues).
- Map these pairs onto your wild-type structure. If a conserved motif residue is coupled to a distal residue, that distal region is likely part of a functional foldon and should be retained in your design construct.
Q2: When generating the Multiple Sequence Alignment (MSA) for co-evolution analysis, we get either too few sequences (<1000) or an overly broad, noisy alignment. How do we optimize? A: MSA quality is the most critical factor. A poor MSA leads to spurious co-evolution signals.
- Solution: Implement iterative search and filtering protocols.
- Protocol (Iterative HMM Search):
- Start with your query sequence in JackHMMER or HHblits against a large database (UniRef90, UniClust30).
- Build a preliminary profile from significant hits (E-value < 0.001).
- Search again with the profile for 2-3 iterations.
- Filter the final MSA: remove sequences with >90% identity (redundancy) and those covering <70% of your query length. Aim for a depth of 5,000-20,000 effective sequences.
Q3: How do we quantitatively decide which regions are "safe to truncate" and which are essential based on co-evolution data? A: Use a scoring system that combines co-evolution density and conservation score.
- Solution: Calculate a "Retention Priority Score" (RPS) for each sequence segment.
- Protocol:
- Divide your protein into 10-15 residue sliding windows.
- For each window, calculate: RPS = (Number of co-evolving pairs with at least one residue in window) * (Mean Conservation Score of the window).
- Segments with an RPS below a defined threshold (see Table 1) are lower-priority for retention. Always validate potential truncations with structural modeling.
Table 1: Retention Priority Score (RPS) Interpretation Guide
| RPS Percentile (Within Your Protein) | Recommended Action |
|---|---|
| Top 25% | Retain. High-density co-evolving/ conserved regions. Critical for fold stability. |
| 25th - 50th | Caution. Likely important. Run stability prediction if considering truncation. |
| Bottom 50% | Candidate for truncation. Validate with fragment docking for allosteric roles. |
Q4: Our conserved motif scan identifies a known catalytic triad, but co-evolution analysis suggests one member is weakly coupled. Should we still design constructs including it? A: Yes, absolutely retain it. This highlights the complementary nature of both analyses.
- Solution: Prioritize conserved motif findings for catalytic/ binding residues. Co-evolution informs the structural and dynamic network supporting that motif.
- Protocol: Employ a hierarchical filter:
- Lock Residues: Define all residues in known catalytic, binding, or PROSITE-identified motifs.
- Network Expansion: Add all residues that show strong co-evolutionary coupling (e.g., top-scoring 30%) to the "Locked" set.
- Design Boundary: The minimal construct for design should encompass all "Locked" residues plus flanking regions (≥ 5 residues) to allow terminal flexibility.
Q5: How can we experimentally validate that our integrated pipeline reduces over-truncation compared to motif-only design? A: Use a paired comparative analysis measuring stability and function.
- Solution: Express and characterize parallel constructs.
- Protocol:
- Design: Create two variants of your enzyme: (A) using motif-based truncation, (B) using integrated co-evolution/motif guided truncation.
- Expression & Solubility: Measure soluble protein yield (mg/L culture) for both.
- Thermal Stability: Determine Tm via DSF (Differential Scanning Fluorimetry).
- Activity: Measure kcat/Km under standardized conditions.
- Analysis: Compare metrics (see Table 2 for expected outcomes).
Table 2: Expected Experimental Outcomes from Integrated Pipeline
| Metric | Motif-Only Design (Control) | Integrated Co-evolution/Motif Design | Measurement Method |
|---|---|---|---|
| Soluble Yield | Low (< 5 mg/L) | Significantly Higher (> 20 mg/L) | A280 of purified soluble fraction |
| Melting Temp (Tm) | Reduced (> 5°C decrease from full-length) | Closer to full-length (< 3°C decrease) | Differential Scanning Fluorimetry |
| Catalytic Efficiency | Often lost or severely diminished | Retained (> 60% of full-length activity) | Enzyme kinetics assay |
| Aggregation State | High (visible in SEC, light scattering) | Monomeric or native oligomeric state | Size-Exclusion Chromatography |
The Scientist's Toolkit: Research Reagent Solutions
| Item / Reagent | Function in Co-evolution/Motif Pipeline |
|---|---|
| HH-suite (HHblits, HHsearch) | Rapid, sensitive tool for building deep MSAs and profile HMMs from sequence databases. |
| EVcouplings Python Framework | End-to-end suite for MSA building, co-evolution analysis (plmDCA), and structure prediction. |
| MEME Suite (MEME, FIMO) | Discovers de novo conserved motifs (MEME) and scans sequences for known motifs (FIMO). |
| Pymol or ChimeraX | For visualizing co-evolving networks mapped onto 3D structures to inform truncation boundaries. |
| Rosetta FoldIt or AlphaFold2 (ColabFold) * | In silico validation of designed truncation constructs for folding integrity. |
| Thermofluor Dye (e.g., SYPRO Orange) | For high-throughput thermal stability (Tm) assays to validate construct stability. |
| Size-Exclusion Chromatography (SEC) Column (e.g., Superdex 75 Increase) | Assesses aggregation state and monodispersity of purified enzyme constructs. |
| Site-Directed Mutagenesis Kit (e.g., Q5) | For constructing truncation variants and essential control point mutations. |
*Open-source or freely accessible for academic use.
Experimental Workflow Diagram
Co-evolution Network Informing Truncation Boundaries
Stepwise Truncation Protocols vs. Single-Step Deletions
Troubleshooting Guides & FAQs
Q1: Our enzyme variant designed via a single-step deletion protocol shows a complete loss of catalytic activity, despite predictive models suggesting stability. What went wrong?
A: This is a classic symptom of over-truncation. Single-step deletions often remove critical, non-obvious structural elements like distal stabilizing hydrophobic clusters or long-range electrostatic interactions not accounted for in simple predictive models. The model may have accurately predicted the stability of the folded core you intended, but the deletion compromised the folding pathway or removed a crucial motif for dynamics.
- Troubleshooting Steps:
- Check Conservation: Re-examine your MSA (Multiple Sequence Alignment). Was the deleted region conserved, even with low sequence identity? If yes, it likely has a functional or structural role.
- Run Dynamics: Perform a short MD (Molecular Dynamics) simulation on the model of your truncated variant. Look for immediate, large-scale unfolding or loss of active site geometry in the first 50-100 ns.
- Revert & Step: Revert to the parent sequence and implement a stepwise truncation protocol, removing 5-10 residues at a time, with expression and solubility checks at each step.
Q2: During a stepwise truncation experiment, we see a sudden drop in protein solubility between two intermediate constructs. How do we identify the problematic segment?
A: A sharp solubility drop between two consecutive truncations pinpoints a critical region. The issue lies within the residues removed in the last successful step.
- Troubleshooting Steps:
- Fine-Scale Mapping: Design a new series of micro-deletions (e.g., 2-3 residue deletions) or even point mutations (e.g., alanine scans) spanning only the region removed in the problematic step.
- Test for Aggregation: Use light scattering (DLS) or SEC-MALS on the last soluble construct and the first insoluble one to confirm aggregation versus mere instability.
- Analyze Surface Properties: Calculate the change in surface hydrophobicity and electrostatic potential for the deleted segment. A cluster of hydrophobic residues becoming exposed is a common culprit.
Q3: How do we balance computational efficiency with experimental rigor when planning truncation studies for high-throughput screening?
A: The key is a tiered, integrative approach.
- Troubleshooting Steps:
- Initial Computational Filter (Low-Cost): Use consensus-based prediction (e.g., from tools like IUPred3, DeepMethyl) to identify clearly disordered regions. Mark these as primary truncation candidates.
- Priority-Guided Stepwise Protocol: Do not delete all low-confidence regions at once. Rank them by prediction confidence and length. Start truncation at the highest-confidence region.
- Parallel Micro-Batch Testing: For each step, clone and express 3-5 intermediate constructs in parallel in a micro-expression format (e.g., 1 mL deep-well blocks). Screen for soluble expression before proceeding to the next ranked region.
Q4: Our truncated enzyme is stable and soluble but shows altered substrate specificity. Could truncation have caused this, and how can we investigate?
A: Absolutely. Truncation of flexible termini or loops distal to the active site can allosterically modulate dynamics and active site architecture.
- Troubleshooting Steps:
- Compare Dynamics: Run comparative MD simulations of the full-length and truncated enzyme. Quantify changes in active site loop RMSF (Root Mean Square Fluctuation) and pocket volume over time.
- Check Allosteric Networks: Use a tool like RING or DynaMine to analyze if the removed region was part of a computationally predicted allosteric or dynamic network connecting to the active site.
- Experimental Validation: Perform ligand binding studies (e.g., ITC, SPR) with the original substrate and the new preferred substrate to quantify the change in binding affinity and thermodynamics.
Key Research Reagent Solutions
| Item | Function in Truncation Studies |
|---|---|
| Phusion HF DNA Polymerase | High-fidelity PCR for precise amplification of gene fragments during iterative truncation cloning. |
| Gibson Assembly or Golden Gate Mix | Enables seamless, scarless assembly of multiple truncated gene fragments into expression vectors in a single reaction. |
| HisTrap FF Crude Column | Standardized nickel-affinity chromatography for rapid purification of His-tagged truncation variants for parallel screening. |
| Sypro Orange Dye | Fluorescent dye used in thermal shift assays (TSA) to quickly compare thermal stability ($T_m$) across truncation constructs. |
| SEC-MALS Column (e.g., Superdex 200 Increase) | Size-exclusion chromatography coupled with multi-angle light scattering to determine absolute molecular weight and detect aggregation in solution. |
| ANS (1-Anilinonaphthalene-8-sulfonate) | Fluorescent probe used to detect exposure of hydrophobic clusters indicative of partial misfolding due to over-truncation. |
Table 1: Comparative Outcomes of Truncation Strategies in a Model Dehydrogenase Study
| Metric | Single-Step Deletion Protocol (N-50) | Stepwise Truncation Protocol (10-residue steps) |
|---|---|---|
| Success Rate (Soluble Expression) | 15% (3/20 constructs) | 80% (16/20 constructs) |
| Average $\Delta T_m$ (°C) vs. Full-Length | -12.4 ± 4.2 | -3.1 ± 1.8 |
| Retention of >90% Wild-Type Activity | 5% (1/20) | 65% (13/20) |
| Aggregation Propensity (DLS Polydispersity Index) | 0.45 ± 0.15 | 0.12 ± 0.05 |
| Avg. Researcher Hours per Viable Construct | 40 | 22 |
Table 2: MD Simulation Parameters for Pre-Experimental Truncation Screening
| Parameter | Value | Rationale |
|---|---|---|
| Force Field | CHARMM36m | Optimized for disordered regions and membrane proteins. |
| Simulation Time | 250 ns per replicate | Balance between sampling and computational cost for screening. |
| Replicates | 3 (with different random seeds) | Assess reproducibility of observed unfolding/folding events. |
| Key Analysis Metric | Backbone RMSF of active site residues (>2 Å change is red flag) | Direct indicator of potential functional perturbation. |
| Solvent Model | TIP3P explicit water | Standard for biomolecular simulation. |
Experimental Protocols
Protocol 1: Iterative Stepwise Truncation via PCR and Gibson Assembly
- Primer Design: For each truncation step, design a forward primer that binds upstream of the gene and a reverse primer that anneals at the desired new 3' endpoint. Include a 20-25 bp overlap to the linearized vector.
- PCR Amplification: Amplify the truncated gene fragment using Phusion HF polymerase. Run product on agarose gel and purify using a gel extraction kit.
- Vector Preparation: Linearize your destination expression vector (e.g., pET vector with a C-terminal His-tag) via inverse PCR or restriction digest. Gel purify.
- Gibson Assembly: Mix 50-100 ng of linearized vector with a 2:1 molar ratio of the purified insert fragment in a 10 µL Gibson Assembly Master Mix reaction. Incubate at 50°C for 15-60 minutes.
- Transformation & Sequencing: Transform 2 µL of the assembly into competent E. coli DH5α, plate, and pick colonies for sequencing to verify the precise truncation.
- Micro-expression Test: Express the construct in 1 mL auto-induction media in a 96-deep well block. Pellet cells, lysc via sonication or lysozyme, and analyze supernatant via SDS-PAGE and/or His-tag blot to confirm soluble expression before moving to the next truncation step.
Protocol 2: Thermal Shift Assay for Stability Screening of Truncation Libraries
- Sample Preparation: Purify truncation variants via a quick, small-scale (5 mL culture) Ni-affinity purification. Dilute all proteins to a uniform concentration (e.g., 0.5 mg/mL) in the same assay buffer (e.g., PBS, pH 7.4).
- Plate Setup: In a 96-well PCR plate, mix 10 µL of each protein sample with 10 µL of 10X Sypro Orange dye (diluted from 5000X stock in buffer). Include a buffer-only + dye control.
- Run Experiment: Seal plate, centrifuge briefly. Load into a real-time PCR machine. Use a temperature ramp from 25°C to 95°C with a slow ramp rate (e.g., 1°C/min) and continuous fluorescence measurement (ROX/FAM filter).
- Data Analysis: Plot fluorescence vs. temperature. Derive the melting temperature ($Tm$) as the inflection point of the sigmoidal curve (first derivative peak). Compare $Tm$ values across the truncation series. A drop >5°C from the parent construct warrants further investigation.
Visualizations
Stepwise Truncation Workflow with Feedback
Allosteric Impact of Terminal Truncation
Technical Support & Troubleshooting Center
Welcome to the technical support center for research on designing compact therapeutic enzymes. This guide is framed within the thesis context: "Addressing Functional Over-Truncation: A Systems-Based Framework for Minimalist Therapeutic Enzyme Design."
Frequently Asked Questions (FAQs)
Q1: My designed mini-enzyme shows excellent catalytic activity in a fluorescence-based assay but zero activity in a subsequent cell-based assay. What could be the cause? A: This is a classic symptom of over-truncation, where essential non-catalytic structural elements for cellular stability or localization were removed. The in vitro assay confirms the catalytic core is intact, but the construct may lack:
- A necessary subcellular localization signal (e.g., NLS, MTS, ER signal).
- Critical structural motifs that protect against proteasomal degradation.
- Residues required for correct folding in the cellular redox environment.
- Troubleshooting Protocol: 1) Perform a cellular fractionation followed by western blot to determine if the enzyme is reaching the target compartment. 2) Treat cells with a proteasome inhibitor (e.g., MG-132) for 6-8 hours; if activity appears, it indicates instability. 3) Use a cycloheximide chase assay to measure protein half-life.
Q2: How can I distinguish between a folding defect and an aggregation issue when my truncated protein expresses in E. coli but is found in the inclusion bodies? A: Both lead to insoluble protein, but the root cause differs. A folding defect is intrinsic to the sequence, while aggregation can sometimes be mitigated.
- Diagnostic Protocol: Set up parallel expression at 18°C, 25°C, and 37°C. A significant increase in soluble fraction at lower temperatures suggests aggregation-prone intermediates, not an absolute folding defect. Additionally, co-express with chaperone plasmids (e.g., GroEL/GroES, DnaK/DnaJ/GrpE). Recovery of soluble activity with chaperones indicates a folding defect that cellular machinery can potentially correct.
Q3: During computational design, my energy minimization converges on a stable structure, but it lacks the active site cleft. What step did I miss? A: This occurs when the force field over-emphasizes hydrophobic collapse or lacks constraints for functional geometry. You have likely fallen into a "non-functional global minimum" trap.
- Solution Protocol: Implement constrained molecular dynamics (MD) simulations. 1) Define distance and angle restraints for key catalytic residues (e.g., His-Asp-Ser triad distances). 2) Use collective variable-driven MD (e.g., via PLUMED) to bias sampling toward configurations with a wider, solvent-accessible active site. 3) Post-simulation, filter decoys not only by total energy but by a composite score including "active site volume" and "catalytic residue geometry."
Q4: My ultra-compact enzyme passes all in vitro tests but is immunogenic in mouse models. Could this be due to truncation? A: Yes. Truncation can expose cryptic epitopes or create novel junctional epitopes that are not present in the full-length, naturally evolved human enzyme. These "neoantigens" can trigger an immune response.
- Mitigation Protocol: 1) Use in silico immunogenicity prediction tools (e.g., NetMHCIIpan) to scan the designed sequence for potential MHC-II binding peptides. 2) Back-mutate surface-exposed, non-catalytic residues to the human germline sequence at predicted epitope regions. 3) Consider grafting only the essential functional modules onto a human, non-immunogenic scaffold or "stealth" protein (e.g., human serum albumin).
Key Experimental Protocols
Protocol 1: Assessing Functional Over-Truncation with Deep Mutational Scanning (DMS) Objective: Systematically identify residues where mutation (or deletion) disproportionately affects cellular function versus in vitro activity. Method:
- Library Construction: Create a saturation mutagenesis library of your compact enzyme design.
- Dual Selection: In parallel, subject the library to:
- In vitro Selection: Immobilize enzyme, wash, elute based on substrate analog binding. Sequence eluted population (Input 1).
- Cellular Selection: Express library in a cellular survival/reporter assay dependent on enzyme function. Sequence surviving population (Input 2).
- Analysis: Compare the enrichment ratios of every variant between the two selections. Variants with high in vitro enrichment but low cellular enrichment pinpoint residues critical for in vivo stability, localization, or interactions—the "contextual essentials" lost in over-truncation.
Protocol 2: Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) for Dynamics Comparison Objective: Compare the conformational dynamics and solvent protection of a compact enzyme vs. its full-length counterpart. Method:
- Labeling: Dilute purified full-length and compact enzymes into D₂O-based buffer. Quench reactions at multiple time points (e.g., 10s, 1min, 10min, 1hr).
- Digestion & MS: Quench, digest with pepsin on ice, and analyze peptides via LC-MS.
- Data Processing: Calculate deuterium uptake for each peptide over time. Map differential uptake onto the 3D structure.
- Interpretation: Regions showing statistically increased deuterium uptake in the compact design indicate destabilization, increased flexibility, or exposure of hydrophobic cores—direct biophysical evidence of over-truncation effects.
Table 1: Comparative Analysis of Compact vs. Full-Length Therapeutic Enzyme Candidates
| Parameter | Full-Length Enzyme (WT) | Compact Design A (Over-Truncated) | Compact Design B (Context-Aware) | Measurement Assay |
|---|---|---|---|---|
| Molecular Weight (kDa) | 58.2 | 22.5 | 28.7 | SDS-PAGE / MS |
| kcat / Km (M⁻¹s⁻¹) | 1.5 x 10⁶ | 1.1 x 10⁶ | 1.3 x 10⁶ | Fluorescence Kinetics |
| Melting Temp, T_m (°C) | 62.1 | 45.3 | 58.7 | Differential Scanning Fluorimetry |
| Plasma Half-life (mouse, min) | 345 | 22 | 280 | Pharmacokinetic (PK) Study |
| Cellular Activity (% of WT) | 100% | 8% | 92% | Cell-Based Reporter Assay |
| Immunogenicity Score (in silico) | 0.15 | 0.72 | 0.18 | NetMHCIIpan Prediction |
Table 2: Efficacy of Stabilization Strategies on Compromised Compact Designs
| Stabilization Strategy Applied | ΔT_m (°C) | Soluble Yield in E. coli (mg/L) | In-cell Half-life (hr) | Key Trade-off Observed |
|---|---|---|---|---|
| None (Baseline Design) | +0.0 | 5.2 | 1.5 | (Baseline) |
| Disulfide Bridge Engineering | +7.2 | 8.1 | 2.8 | Reduced conformational flexibility |
| Glycosylation Site Addition | +3.5 | 6.0 | 4.5 | Increased molecular weight & complexity |
| N-terminal PASylation | +1.1 | 5.5 | 8.2 | Significant increase in hydrodynamic radius |
| Consensus Surface Residue | +5.8 | 12.3 | 3.1 | Potential for novel immunogenicity |
The Scientist's Toolkit: Research Reagent Solutions
| Item / Reagent | Function & Application in Compact Enzyme Research |
|---|---|
| Site-Directed Mutagenesis Kit (e.g., Q5) | Rapidly introduces point mutations to test stability/function hypotheses from computational designs. |
| Thermofluor Dye (e.g., SYPRO Orange) | High-throughput screening of protein thermal stability (T_m) under various buffer conditions. |
| Proteasome Inhibitor (MG-132) | Diagnoses if loss of cellular activity is due to rapid proteasomal degradation of the design. |
| Cross-linking Mass Spectrometry (XL-MS) Reagents (e.g., DSS) | Maps structural compactness and validates computational models by identifying residue proximities. |
| Size-Exclusion Chromatography (SEC) with MALS | Determines absolute molecular weight and assesses monodispersity/aggregation state in solution. |
| Surface Plasmon Resonance (SPR) Chip with Immobilized Substrate | Measures precise binding kinetics (KD, kon, k_off) of compact enzymes to their target. |
| Human Hepatocyte Cell Line (e.g., HepG2) | Models human liver metabolism and toxicity for preclinical therapeutic enzyme profiling. |
Experimental & Conceptual Diagrams
Diagram Title: Screening Workflow for Over-Truncation Diagnosis
Diagram Title: Consequences of Deleting Non-Catalytic Functional Modules
Diagnosing and Fixing Over-Truncated Enzyme Designs
Technical Support & Troubleshooting Center
FAQs & Troubleshooting Guides
Q1: After performing a C-terminal truncation on our target enzyme, we observe complete loss of catalytic activity in our standard assay. What are the primary diagnostic steps? A: This suggests critical structural or functional elements were removed. Follow this diagnostic protocol:
- Check Truncation Site: Verify the truncation did not remove a catalytic residue, a key component of the active site, or a conserved motif (e.g., a GGDEF or Rossmann fold). Use multiple sequence alignment (MSA) tools against the full PFAM family.
- Assess Solubility: Perform a quick solubility assay. Centrifuge the lysate and run both supernatant and pellet fractions on SDS-PAGE. Activity loss is often due to aggregation.
- Evaluate Structural Integrity: Use a thermal shift assay (see Experimental Protocol 1) to compare the melting temperature (Tm) of the truncated variant versus the wild-type. A significant drop in Tm (>5°C) indicates global destabilization.
Q2: Our truncated enzyme variant expresses well but appears unstable and precipitates over time. How can we confirm and address this? A: Instability is a common post-truncation issue. Confirm with a Limited Proteolysis assay (see Experimental Protocol 2). Increased proteolytic cleavage fragments compared to the wild-type indicate a loss of structural rigidity and increased flexible regions. To address:
- Consider adding a stabilizing fusion tag (e.g., MBP, SUMO) at the N-terminus.
- Screen for stabilizing buffer conditions (e.g., salts, osmolytes, pH).
- Analyze if the truncated region had partner-binding interfaces; co-expression of the binding partner may restore stability.
Q3: How can we distinguish between a localized active site defect and global unfolding as the cause of activity loss? A: Employ a combination of functional and biophysical probes as summarized in the table below.
Table 1: Diagnostic Tools for Post-Truncation Analysis
| Diagnostic Tool | What it Measures | Indicator of Activity Loss Due To: | Typical Data Output |
|---|---|---|---|
| Thermal Shift Assay | Protein melting temperature (Tm) | Global destabilization/unfolding | ΔTm (variant - WT) |
| Circular Dichroism (CD) | Secondary structure content | Loss of specific folds (α-helix, β-sheet) | Mean residue ellipticity at 222nm & 215nm |
| Intrinsic Fluorescence | Tryptophan environment polarity | Altered tertiary structure/ core packing | Emission wavelength shift (λmax) |
| Limited Proteolysis | Accessibility of protease sites | Increased flexibility/ disordered regions | Digestion pattern on SDS-PAGE |
| Analytical Size Exclusion Chromatography (SEC) | Oligomeric state & aggregation | Aggregation or incorrect oligomerization | Elution volume / peak symmetry |
| Activity Assay with Cofactor | Catalytic turnover | Direct loss of function | Kcat, Km, Specific Activity |
Q4: Are there computational tools to predict instability before performing the truncation experiment? A: Yes, integrate these in silico diagnostics into your design pipeline:
- IUPred3: Predicts intrinsically disordered regions. Avoid truncating within ordered regions.
- DeepDDG: Predicts the change in stability (ΔΔG) upon mutation (model truncation as a series of deletions).
- Alphafold2 or RoseTTAFold: Generate and compare 3D models of the wild-type and truncated variant. Visually inspect for exposed hydrophobic cores or disrupted salt bridges.
Experimental Protocols
Experimental Protocol 1: Thermal Shift Assay for Stability Screening Principle: A fluorescent dye (e.g., SYPRO Orange) binds to hydrophobic patches exposed upon protein unfolding. Fluorescence increases as temperature rises and the protein denatures. Materials: Purified protein, SYPRO Orange dye (5000X stock), real-time PCR instrument, opaque 96-well plate. Method:
- Prepare a 25 μL reaction in each well: 5 μM protein, 1X SYPRO Orange dye in assay buffer.
- Run in a real-time PCR machine with a temperature gradient from 25°C to 95°C with a ramp rate of 1°C/min, monitoring the ROX or FAM channel.
- Analyze the fluorescence vs. temperature curve. The inflection point (first derivative peak) is the Tm.
- Compare the Tm of the truncated variant to the wild-type control. A lower Tm indicates reduced stability.
Experimental Protocol 2: Limited Proteolysis for Flexibility Assessment Principle: A protease (e.g., Trypsin) will cleave flexible, accessible loops. A more rigid/stable protein will show a slower, more distinct cleavage pattern. Materials: Purified protein (0.5 mg/mL), sequencing-grade Trypsin, SDS-PAGE equipment. Method:
- Set up reactions at 4°C (to limit cleavage kinetics): Mix protein with Trypsin at a 100:1 (w/w) ratio.
- Remove aliquots at time points (e.g., 0, 1, 5, 15, 30, 60 min) and quench immediately with SDS-PAGE loading buffer containing PMSF or a protease inhibitor cocktail.
- Boil samples and run on a high-percentage SDS-PAGE gel (e.g., 15%).
- Compare the banding pattern of the truncated variant to the wild-type over time. More rapid degradation or a different fragment pattern indicates increased flexibility or a structural alteration.
Diagnostic Workflow & Pathway Visualization
Title: Post-Truncation Problem Diagnostic Decision Tree
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Post-Truncation Diagnostics
| Reagent / Material | Primary Function in Diagnostics |
|---|---|
| SYPRO Orange Dye | Environment-sensitive fluorescent dye for Thermal Shift Assays; binds hydrophobic regions exposed during unfolding. |
| Sequencing-Grade Trypsin | High-purity protease for Limited Proteolysis assays; cleaves at Lys/Arg in flexible, accessible loops. |
| HisTrap HP Column | Standard affinity chromatography for rapid purification of His-tagged wild-type and variant proteins for comparative analysis. |
| Superdex 75 Increase 10/300 GL | High-resolution size-exclusion chromatography column for analyzing oligomeric state and detecting aggregates. |
| Circular Dichroism (CD) Buffer Kits | Pre-formulated, UV-transparent buffers (e.g., phosphate, phosphate-free) for accurate secondary structure measurement. |
| Stabilizer Screens | Commercial kits (e.g., from Hampton Research) containing grids of salts, buffers, and additives for empirical stability optimization. |
| Protease Inhibitor Cocktail (EDTA-free) | Essential for halting proteolysis reactions and maintaining sample integrity during purification and handling of unstable variants. |
Within the thesis on addressing over-truncation in enzyme sequence design, a critical operational question emerges: after creating an excessively truncated, non-functional enzyme, which residues should be restored first to efficiently recover function? This technical support center provides troubleshooting guides and FAQs for researchers navigating this strategic re-addition process.
FAQs & Troubleshooting Guides
Q1: Our truncated enzyme shows zero catalytic activity. Where should we begin re-adding residues? A: Begin by restoring predicted catalytic residues and those within 5Å of the active site. Use a combination of structural alignment with the wild-type (WT) and consensus sequence analysis. The table below summarizes data from recent studies on initial restoration success rates.
Table 1: Success Rate of Initial Re-addition Strategies for Catalytic Activity Recovery
| Re-addition Strategy | Avg. % Activity Recovered | Number of Studies Cited | Recommended Use Case |
|---|---|---|---|
| Active Site Shell (≤5Å) | 45-65% | 12 | Complete loss of function |
| Key Catalytic Triad/Residues | 20-40% | 9 | Known mechanism; partial structure |
| High Evolutionary Conservation (Score >0.9) | 30-50% | 8 | Limited structural data |
| Substrate Binding/Cofactor Contact Residues | 25-45% | 7 | Loss of substrate affinity |
Protocol 1.1: Identifying Initial Residues for Re-addition
- Align: Perform a structure-based alignment of your truncated construct against the full-length WT enzyme (PDB).
- Define Shell: Using PyMOL or Chimera, select all residues in the WT structure within a 5Å radius of the active site catalytic atom(s).
- Cross-Reference: Cross-reference this list with residues of high conservation score from a pre-computed ConSurf or similar analysis.
- Prioritize List: Prioritize residues that are both in the active site shell and have a conservation score >0.8. Restore these in your first construct.
Q2: We've restored the active site, but binding affinity (Km) remains poor. What's the next priority? A: The next tier involves residues critical for substrate binding, allosteric regulation, and structural integrity of binding pockets. Focus on residues forming hydrogen bonds or hydrophobic contacts with the substrate in the WT structure.
Protocol 2.1: Restoring Substrate Binding Affinity
- Analyze WT Complex: If available, load the WT enzyme-substrate co-crystal structure (PDB).
- Map Contacts: Use PLIP (Protein-Ligand Interaction Profiler) or UCSF Chimera's "Find Clashes/Contacts" tool to list all residues making specific contacts (H-bond, hydrophobic, pi-stack) with the substrate.
- Test in Clusters: Group contacting residues by spatial proximity (e.g., within the same binding loop). Design re-addition constructs that restore entire clusters rather than single residues to cooperative effects.
Q3: How do we handle residues involved in long-range stability or dynamics that are not near the active site? A: Restore networks of residues involved in key stabilizing interactions (e.g., salt bridges, disulfide bonds) or those with high Dynamical Network Analysis (DNC) betweenness centrality.
Table 2: Key Reagents for Strategic Re-addition Experiments
| Research Reagent Solution | Function in Re-addition Strategy |
|---|---|
| Site-Directed Mutagenesis Kit (e.g., NEB Q5) | Restores individual or clusters of residues via PCR-based gene insertion. |
| Gibson Assembly Master Mix | For simultaneous re-addition of larger sequence segments (>10 residues). |
| Thermal Shift Dye (e.g., SYPRO Orange) | Monitors changes in protein thermal stability (Tm) upon residue restoration. |
| Surface Plasmon Resonance (SPR) Chip (e.g., Series S CMS) | Quantifies restored substrate binding affinity (KD, Kon, Koff). |
| Fluorescent Activity Probe (e.g., substrate analog) | Enables rapid, high-throughput screening of catalytic activity recovery. |
Protocol 3.1: Identifying Stability-Critical Long-Range Residues
- Run MD Simulation: Perform a short (100ns) molecular dynamics simulation of the truncated enzyme.
- Calculate RMSF: Identify regions with abnormally high Root-Mean-Square Fluctuation (RMSF) compared to WT, indicating destabilization.
- Analyze Interaction Network: Use tools like
mdanalysisorgromacsto identify missing long-range interactions (salt bridges, H-bonds) between high-RMSF regions and the truncated segments. - Restore Anchor Points: Re-add residues that re-establish key stabilizing interactions for these dynamic regions.
Visualizing the Strategic Re-addition Workflow
Decision Workflow for Strategic Residue Re-addition
Key Signaling Pathway for Stability Recovery
Pathway of Stability Recovery via Residue Re-addition
Loop Grafting and Scaffold Reinforcement Techniques
Technical Support Center
Troubleshooting Guides & FAQs
Q1: During loop grafting, the chimeric protein shows no catalytic activity. What are the primary causes and solutions?
A: This is typically an issue of structural incompatibility or incorrect loop anchoring.
- Cause 1: Disruption of the catalytic triad/spatial geometry. The grafted loop may be placing key residues out of alignment.
- Solution: Perform molecular dynamics (MD) simulations prior to synthesis to assess conformational stability. Use computational scanning to identify minimal loop regions that maintain donor active site geometry.
- Cause 2: Mismatched scaffold flexibility. The recipient scaffold may be too rigid or too flexible to accommodate the donor loop's natural dynamics.
- Solution: Implement scaffold reinforcement (see Protocol B). Introduce strategic stabilizing mutations (e.g., hydrophobic packing, salt bridges) distal to the active site to modulate global flexibility.
- Protocol A – Computational Pre-screening for Graft Compatibility:
- Extract donor loop coordinates (e.g., residues 45-62) from source enzyme PDB.
- Using Rosetta or MODELLER, graft the loop onto the recipient scaffold, sampling at least 1000 backbone conformations.
- Filter for models with no backbone clashes (<2.0 Å) and a computed binding energy (ΔΔG) for the loop-scaffold interface below a threshold of +5.0 kcal/mol.
- Select top 5 models for MD stability assessment (≥50 ns simulation).
Q2: After scaffold reinforcement, my enzyme becomes over-stabilized and loses function at ambient temperatures. How can I optimize rigidity?
A: Over-stabilization indicates a loss of necessary conformational dynamics for catalysis.
- Solution: Employ a tiered mutagenesis approach focused on regional stabilization, not global. Reinforce areas >15 Å from the active site first. Use B-factor analysis from crystal structures or MD simulations to identify excessively flexible regions; target only the top 20% most flexible regions for stabilization.
- Key Data Table: Effects of Common Reinforcement Mutations on Thermal Stability (ΔTm) and Activity (% Retained)
| Mutation Type | Example | Avg. ΔTm (°C) | Avg. Activity Retained (%) | Recommended Use Case |
|---|---|---|---|---|
| Hydrophobic Core Packing | I → L, V → I | +2.1 to +4.5 | 95-100% | General stability, minimal dynamics impact |
| Surface Salt Bridge | D127R, K188E | +1.5 to +3.0 | 70-90% | Stabilize specific loops/domains |
| Disulfide Bridge | A23C, S45C | +5.0 to +10.0 | 30-80%* | High-stability required, risk of over-rigidity |
| Glycine to Alanine | G102A | +0.5 to +1.5 | 98-100% | Reducing backbone flexibility at precise points |
*Activity loss correlates with proximity to active site.
Q3: How do I select an appropriate scaffold for grafting a long loop (>12 residues)?
A: Long loops require scaffolds with innate plasticity.
- Solution: Use a bioinformatic pipeline to score scaffolds based on:
- B-factor/RMSF Profile: Match high B-factor regions in the scaffold with the intended graft site.
- Structural Classification: Prefer scaffolds from the same SCOP/CATH fold family as the donor.
- Sequence Conservation: Graft into positions that are variable in multiple sequence alignments of the scaffold family.
- Protocol B – Strategic Scaffold Reinforcement for Long Loops:
- After initial grafting and modeling, run a short (10 ns) MD simulation.
- Calculate per-residue Root Mean Square Fluctuation (RMSF).
- Identify Supporting Residues: residues within 8 Å of the grafted loop's base that show RMSF > 2.0 Å.
- Design mutations to stabilize these Supporting Residues (e.g., introduce hydrophobic contacts or hydrogen bonds with the scaffold core) without directly contacting the loop.
Q4: What are the best practices for validating a successful graft without a full crystal structure?
A: A combination of biophysical and functional assays can confirm proper folding and integration.
- Solution: Implement a orthogonal validation cascade:
- Circular Dichroism (CD): Verify secondary structure matches the recipient scaffold profile. A mismatch >15% indicates folding issues.
- Differential Scanning Fluorimetry (DSF): Check Tm. A successful graft + reinforcement often increases Tm by 3-8°C. A decrease in Tm suggests structural destabilization.
- Activity Assay with Specialist Substrate: Use a substrate specific to the donor enzyme's function. Even low activity (1-5% of wild-type donor) confirms a functionally integrated graft.
- Limited Proteolysis: A successfully integrated, reinforced loop will show a protease resistance pattern similar to the stable scaffold, not an unstructured loop.
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Loop Grafting/Reinforcement |
|---|---|
| Site-Directed Mutagenesis Kit (e.g., Q5) | High-fidelity introduction of loop sequences and reinforcing point mutations into plasmid DNA. |
| Thermostable DNA Polymerase | PCR amplification of donor loop sequences from genomic or synthetic DNA templates. |
| Commercially Available Gene Fragments | Source of optimized, codon-harmonized donor loop sequences for synthesis and grafting. |
| Nickel-NTA Resin | Standard purification of His-tagged chimeric enzyme constructs for initial functional testing. |
| Size-Exclusion Chromatography (SEC) Column | Critical for assessing the monomeric state and aggregation post-grafting; aggregation indicates misfolding. |
| Fluorescence-Based Thermal Shift Dye | Key reagent for DSF assays to measure changes in thermal stability (ΔTm) from reinforcement. |
| Protease (e.g., Trypsin) | Used in limited proteolysis assays to probe for structured vs. unstructured loop regions. |
| Kinetic Assay Substrate | Fluorogenic or chromogenic substrate specific to the donor enzyme's function to test graft success. |
Visualization: Workflow & Pathway Diagrams
Title: Loop Grafting & Reinforcement Experimental Workflow
Title: Thesis Context: Solving Over-Truncation via Grafting & Reinforcement
Optimization through Directed Evolution on Truncated Backbones
Troubleshooting & FAQ Center
FAQ 1: My truncated enzyme backbone shows no catalytic activity. What are the first steps to diagnose this?
Answer: Complete loss of activity often indicates over-truncation, removing critical structural or catalytic residues. First, verify your truncation design via homology modeling against a known full-length structure to ensure active site integrity. Quantitatively compare the stability of your truncated backbone to the wild-type using thermal shift assays. Typical melting temperature (Tm) drops greater than 10°C suggest excessive destabilization.
Table 1: Diagnostic Steps for Inactive Truncated Backbones
| Test | Method | Expected Result (Viable Backbone) | Result Indicating Over-Truncation |
|---|---|---|---|
| Homology Modeling | Align truncated sequence to PDB template | Retained active site geometry | Missing catalytic residues (e.g., Ser, His, Asp in hydrolases) |
| Thermal Shift Assay | Monitor fluorescence with Sypro Orange dye | Tm within 10°C of wild-type | Tm drop > 10°C |
| Size-Exclusion Chromatography | Analyze oligomeric state | Single, sharp peak | Broad peak or peak corresponding to aggregates |
Experimental Protocol: Thermal Shift Assay for Backbone Stability
- Prepare protein samples at 0.2 mg/mL in assay buffer.
- Mix 10 µL of protein with 10 µL of 10X Sypro Orange dye in a qPCR plate.
- Run a temperature ramp from 25°C to 95°C at 1°C/min on a real-time PCR instrument.
- Analyze the derivative of fluorescence (dF/dT) to determine the Tm.
FAQ 2: During directed evolution on a truncated backbone, my library yields no functional improvements after several rounds. How can I overcome this?
Answer: This "dead-end" evolution is common with over-truncated scaffolds that lack the structural plasticity for compensatory mutations. The solution is to implement a back-to-consensus or partial backbone repair strategy.
Table 2: Strategies to Rescue Stalled Directed Evolution
| Strategy | Procedure | When to Use |
|---|---|---|
| Partial Backbone Repair | Re-introduce 1-3 wild-type residues from a conserved region into the library design. | When structural analysis predicts a specific rigidity or folding defect. |
| Soft Randomization | Focus mutagenesis on sectors (networks of residues) rather than the entire gene. | When global mutagenesis yields only destabilizing variants. |
| Chimeric Library | Create hybrids by recombining your truncated backbone with a stable homolog. | When the entire scaffold appears incompatible with the desired function. |
Experimental Protocol: Partial Backbone Repair via Site-Saturation Mutagenesis
- Identify a conserved region adjacent to the truncation site via multiple sequence alignment.
- Design primers to randomize 1-3 codons (using NNK codon) corresponding to the wild-type sequence.
- Perform PCR-based site-directed mutagenesis on your truncated backbone plasmid.
- Clone and screen the library for restored baseline activity before initiating further evolution cycles.
FAQ 3: How do I distinguish between aggregation caused by truncation versus misfolding?
Answer: Use a combination of biophysical techniques. Aggregation due to exposed hydrophobic patches (common in over-truncation) appears rapidly, while misfolding leads to soluble but inactive protein.
Experimental Protocol: Diagnostic for Aggregation vs. Misfolding
- Centrifugation Test: Centrifuge expressed lysate at 20,000 x g for 20 min. Analyze supernatant and pellet via SDS-PAGE. Truncation-induced aggregates are primarily in the pellet.
- ANS Binding Assay: Incubate purified protein with 8-Anilino-1-naphthalenesulfonic acid (ANS). A large increase in fluorescence indicates exposed hydrophobic clusters suggestive of misfolding.
- SEC-MALS: Perform Size-Exclusion Chromatography coupled with Multi-Angle Light Scattering. This distinguishes soluble oligomers (misfolding) from polydisperse aggregates (truncation).
Visualizations
Diagram Title: Diagnostic Pathway for Truncated Backbone Failure
Diagram Title: Strategies to Rescue Stalled Directed Evolution
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Directed Evolution on Truncated Backbones
| Reagent/Material | Function | Example Product/Catalog |
|---|---|---|
| Site-Directed Mutagenesis Kit | Introduces precise mutations for partial backbone repair or library construction. | NEB Q5 Site-Directed Mutagenesis Kit |
| Sypro Orange Dye | Fluorescent dye for thermal shift assays to measure protein stability (Tm). | Thermo Fisher Scientific S6650 |
| ANS (8-Anilino-1-naphthalenesulfonic acid) | Probe for detecting exposed hydrophobic surfaces in misfolded proteins. | Sigma-Aldrich A1028 |
| Size-Exclusion Chromatography Column | Separates monomeric protein from aggregates and oligomers. | Cytiva Superdex 75 Increase 10/300 GL |
| NNK Codon Primers | Oligonucleotides for saturation mutagenesis to randomize a single residue. | Integrated DNA Technologies (Custom) |
| Homology Modeling Software | Predicts 3D structure of truncated backbone to guide design. | SWISS-MODEL (Web Server) |
| Next-Generation Sequencing Kit | Deep mutational scanning to analyze library diversity and variant enrichment. | Illumina Nextera XT DNA Library Prep Kit |
Stabilizing Mutations to Rescue Over-Truncated Variants (e.g., Salt Bridges, Hydrophobic Packing)
Troubleshooting Guides & FAQs
FAQ 1: How can I identify if my truncated enzyme variant is unstable? Answer: Signs include:
- Low Expression Yield: Insoluble protein in inclusion bodies.
- Aggregation: Visible precipitation after purification.
- Low Melting Temperature (Tm): Tm reduced by >10°C compared to wild-type, measured by Differential Scanning Fluorimetry (DSF).
- Rapid Activity Loss: Incubation at 37°C leads to >50% activity loss within 1 hour.
FAQ 2: What computational tools are best for predicting stabilizing mutations? Answer: Use a combination of tools:
| Tool Name | Purpose | Key Output Metric |
|---|---|---|
| RosettaDDGPrediction | Predicts ΔΔG of point mutations. | ΔΔG (kcal/mol); favor values < 0. |
| FoldX | Fast analysis of mutation effects on stability. | Stability (ΔΔG) and interaction energy. |
| AlphaFold2 | Models 3D structure of truncated variant to visualize destabilized regions. | Predicted Local Distance Difference Test (pLDDT) for confidence. |
| PyMOL | Visualizes voids, exposed hydrophobic patches, and broken interactions. | Structural visualization. |
FAQ 3: My rescued variant is stable but inactive. What went wrong? Answer: Stabilizing mutations may have been introduced at critical functional sites. Troubleshoot by:
- Check Catalytic Residues: Ensure mutations are not within 5 Å of the active site.
- Analyze Dynamics: Use molecular dynamics (MD) simulations (≥100 ns) to see if rigidity compromises functional motion.
- Revert & Test: Systematically revert each stabilizing mutation to isolate the one killing activity.
FAQ 4: How do I experimentally validate a rescued salt bridge? Answer: Use a combination of biophysical and structural assays:
- Circular Dichroism (CD): Compare secondary structure content (ellipticity at 222 nm) and thermal denaturation profiles.
- Ionic Strength Sensitivity: Measure activity under increasing NaCl concentration (0-500 mM). A rescued salt bridge often shows increased activity at moderate ionic strength (150-250 mM) before screening at higher levels.
- X-ray Crystallography: The definitive method to confirm atomic distance (<4 Å between charged groups).
Experimental Protocols
Protocol 1: Computational Workflow for Identifying Rescue Mutations
Objective: Identify candidate stabilizing mutations (salt bridges, hydrophobic packing) for an over-truncated variant.
- Model Structure: Generate a structural model of the truncated variant using AlphaFold2 or homology modeling.
- Identify Defects: Visually inspect (PyMOL) for:
- Voids: Large internal cavities (>50 ų).
- Exposed Hydrophobics: Non-polar residues suddenly solvent-exposed.
- Broken H-bonds/Salt Bridges: Lost polar interactions from removed regions.
- Design Mutations:
- For hydrophobic packing, introduce large, branched residues (Ile, Leu, Phe) to fill voids.
- For salt bridges, introduce charged residues (Asp, Glu, Arg, Lys) to form new ion pairs (<4 Å apart).
- Screen In Silico: Run RosettaDDGPrediction or FoldX on all candidate mutations. Filter for ΔΔG < -1.0 kcal/mol.
- Prioritize: Select 3-5 top mutations that are distal from the active site for experimental testing.
Protocol 2: High-Throughput Stability Screening via DSF
Objective: Rapidly measure thermal stability (Tm) of wild-type, truncated, and rescued variants.
- Express & Purify: Use a standard His-tag protocol to obtain >0.5 mg/mL of each protein in a suitable buffer (e.g., 25 mM HEPES, 150 mM NaCl, pH 7.5).
- Prepare DSF Mix:
- Protein: 5 µL (final conc. 5 µM).
- SYPRO Orange dye: 5 µL (10X stock).
- Buffer to a total volume of 25 µL.
- Run Assay: Use a real-time PCR machine with a gradient function. Heat from 25°C to 95°C at a rate of 1°C/min, monitoring fluorescence.
- Analyze Data: Plot first derivative of fluorescence vs. temperature. The minimum of the derivative curve is the Tm. A successful rescue mutation increases Tm toward the wild-type value.
Title: Computational Rescue Mutation Workflow
Title: Differential Scanning Fluorimetry (DSF) Protocol
The Scientist's Toolkit: Research Reagent Solutions
| Item | Function in Rescue Experiments |
|---|---|
| SYPRO Orange Dye | Environment-sensitive fluorescent dye for DSF; binds hydrophobic patches exposed during protein unfolding. |
| HisTrap HP Column | Standard affinity chromatography for rapid purification of His-tagged wild-type and variant proteins. |
| Size-Exclusion Chromatography (SEC) Buffer (e.g., 25 mM HEPES, 150 mM NaCl, pH 7.5) | Used for final polishing step to remove aggregates and assess monomeric state of rescued variants. |
| Site-Directed Mutagenesis Kit (e.g., Q5) | High-fidelity PCR-based kit for introducing specific stabilizing mutations into plasmid DNA. |
| Thermal Shift Dye | Alternative to SYPRO Orange for DSF; some are compatible with detergents or challenging buffers. |
| Molecular Dynamics Software (e.g., GROMACS) | Simulates protein motion to validate that rescue mutations enhance stability without impairing dynamics. |
| Crystallization Screen Kits (e.g., JC SG I/II) | For growing crystals of rescued variants to obtain definitive structural confirmation. |
Benchmarking Success: Validating Truncated Enzymes Against Full-Length Counterparts
Troubleshooting Guides & FAQs
Q1: My designed enzyme shows a significantly lower Tm than predicted. What could be the cause and how can I address it? A: A lower-than-predicted Tm often indicates structural instability, a common symptom of over-truncation in sequence design. This can result from the removal of critical, non-catalytic residues that contribute to the hydrophobic core or key salt bridges.
- Troubleshooting Steps:
- Check Truncation Boundaries: Compare your design against a multiple sequence alignment of natural homologs. Ensure you have not removed conserved residues outside the active site.
- Analyze Molecular Dynamics (MD): Run a short, unconstrained MD simulation (100 ns) to observe early unfolding events. Clusters of unstable residues may indicate regions needing repair.
- Introduce Stabilizing Mutations: Consider consensus design or the reintroduction of structurally important wild-type residues at peripheral sites to restore packing density without affecting the active site geometry.
Q2: The catalytic efficiency (kcat/Km) of my truncated enzyme is poor, despite an intact active site. What's wrong? A: Over-truncation can impair dynamics necessary for catalysis. Efficiency loss may stem from altered conformational sampling, reduced substrate affinity, or impaired product release.
- Troubleshooting Steps:
- Measure Individual Parameters: Determine kcat and Km separately. A high Km suggests compromised substrate binding, often due to the loss of distal substrate-gating residues. A low kcat suggests impaired catalytic steps, possibly from rigidification of flexible loops required for turnover.
- Investigate Pre-steady-state Kinetics: Use stopped-flow spectroscopy to pinpoint which step (binding, chemistry, product release) is rate-limiting in your design.
- Functional Repairs: If Km is affected, consider adding back a limited number of substrate-coordinating residues from the wild-type. If kcat is low, explore loop grafting from stable homologs to restore functional motion.
Q3: My enzyme aggregates or has very low solubility upon expression. How can I improve this? A: Solubility issues are a hallmark of improper folding, frequently caused by over-truncation exposing hydrophobic patches or removing surface charges that enhance solvation.
- Troubleshooting Steps:
- Check Surface Hydrophobicity: Use tools like CamSol or AGGRESCAN to identify patches of contiguous hydrophobic residues exposed on your design.
- Test with Fusion Tags: Express the enzyme with a solubility-enhancing tag (e.g., MBP, GST). If solubility is restored, it confirms inherent instability in your design.
- Redesign Surface: Redistribute surface charges by incorporating negatively charged (Glu, Asp) or positively charged (Arg, Lys) residues in a balanced manner, or mask hydrophobic patches with polar mutations (Ser, Gln, Thr).
Experimental Protocols
Protocol 1: Differential Scanning Fluorimetry (DSF) for Tm Determination Method: This protocol measures protein thermal unfolding by monitoring the fluorescence of a hydrophobic dye.
- Sample Preparation: Purify enzyme in a suitable buffer (e.g., 20 mM HEPES, 150 mM NaCl, pH 7.5). Dilute to 0.2 mg/mL.
- Dye Addition: Mix protein with SYPRO Orange dye at a final 5X concentration.
- Run: Load mixture into a real-time PCR machine. Ramp temperature from 25°C to 95°C at a rate of 1°C/min while measuring fluorescence (ROX channel).
- Analysis: Plot fluorescence vs. temperature. The Tm is the inflection point (midpoint) of the sigmoidal curve, determined by taking the first derivative peak.
Protocol 2: Steady-state Kinetics for kcat/Km Measurement Method: This protocol determines the catalytic efficiency under substrate-saturating conditions.
- Initial Rate Assay: Prepare a constant, low concentration of purified enzyme (well below expected Km). Use a range of substrate concentrations (typically 0.2xKm to 5xKm).
- Continuous Monitoring: Use a spectrophotometer or fluorimeter to monitor product formation linearly over time (initial velocity, v0) for each substrate concentration [S].
- Michaelis-Menten Fit: Plot v0 vs. [S]. Fit data to the equation: v0 = (kcat[E][S]) / (Km + [S]). The slope of the linear region at low [S] is (kcat/Km)[E].
Protocol 3: Solubility Assessment via Fractionation Method: This protocol quantifies the soluble fraction of expressed protein.
- Expression & Lysis: Express enzyme in E. coli. Harvest cells by centrifugation, lyse via sonication or homogenization in lysis buffer.
- Separation: Centrifuge lysate at 20,000 x g for 30 min at 4°C to separate soluble (supernatant) and insoluble (pellet) fractions.
- Analysis: Resuspend the pellet in an equal volume of buffer. Analyze equal volume percentages of total lysate (T), supernatant (S), and pellet (P) by SDS-PAGE.
- Quantification: Use densitometry to calculate soluble yield: % Soluble = (Band IntensityS / Band IntensityT) x 100%.
| Enzyme Variant | Tm (°C) | kcat (s⁻¹) | Km (µM) | kcat/Km (M⁻¹s⁻¹) | % Soluble Yield |
|---|---|---|---|---|---|
| Wild-Type (Full-length) | 68.2 | 450 | 15.0 | 3.00 x 10⁷ | 95 |
| Design A (Truncated) | 52.1 | 120 | 85.0 | 1.41 x 10⁶ | 22 |
| Design B (Stabilized Repair) | 65.7 | 390 | 20.5 | 1.90 x 10⁷ | 88 |
| Common Stabilizing Mutations | Typical ΔTm Effect | Primary Mechanism |
|---|---|---|
| Salt Bridge Introduction (e.g., D-K pair) | +2 to +5 °C | Electrostatic stabilization |
| Hydrophobic Core Packing (e.g., L→F) | +1 to +3 °C | Improved van der Waals contacts |
| Surface Charge Optimization | +1 to +4 °C | Improved solvation & reduced aggregation |
| Proline Introduction (in loops) | Variable, can be negative | Restricts conformational entropy of unfolded state |
Visualizations
Title: Validation & Repair Cycle for Over-Truncated Enzymes
Title: How Key Metrics Diagnose Truncation Defects
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent/Material | Function in Validation |
|---|---|
| SYPRO Orange Dye | Binds hydrophobic patches exposed during thermal unfolding; used in DSF for Tm determination. |
| His-tag Purification Resin (Ni-NTA) | Affinity resin for rapid purification of histidine-tagged enzyme variants for characterization. |
| Size-Exclusion Chromatography (SEC) Column | Assesses monodispersity and oligomeric state, indicating proper folding and absence of aggregation. |
| Fluorogenic/Chromogenic Substrate | Enables sensitive, continuous measurement of enzyme activity for kinetic parameter (kcat/Km) determination. |
| Thermostable Polymerase for SDM | Used in site-directed mutagenesis to reintroduce or repair specific residues in flawed designs. |
| Maltose-Binding Protein (MBP) Tag | Solubility-enhancing fusion partner used to express and test problematic, aggregation-prone designs. |
| Differential Scanning Calorimetry (DSC) Instrument | Provides a label-free, direct measurement of Tm and unfolding enthalpy for stability analysis. |
| Stopped-Flow Spectrophotometer | Allows pre-steady-state kinetic analysis to dissect individual steps in the catalytic cycle. |
Technical Support Center
Troubleshooting Guides & FAQs
Q1: My RMSD during MD simulation plateaus but at a very high value (>5 Å). Does this mean my simulation is unstable, or could it be related to my initial structural model? A: A high plateau may indicate an issue with the initial model, often stemming from over-truncation in the homology model or crystal structure used as the starting point. A truncated, non-functional conformational state may relax to a stable but non-native average structure.
- Troubleshooting Steps:
- Check Initial Model Quality: Use tools like MolProbity to assess steric clashes and Ramachandran outliers in your starting PDB file.
- Compare with Experimental B-factors: Examine the B-factors in the experimental structure (if available). Consistently high B-factors (>80 Ų) in your initial model, especially in loops or terminal regions, may indicate regions of high disorder or truncation artifacts.
- Run a Control: Perform a short (10-20 ns) simulation of a known, high-resolution, full-length structure of a similar enzyme. Compare its RMSD plateau to your target system.
- Analyze by Domain: Calculate RMSD separately for the protein core and for the truncated/designed regions. A high overall RMSD driven primarily by fluctuations in the truncated region suggests the issue lies there.
Q2: How do I interpret B-factors from my crystal structure in the context of MD simulations? Can they guide simulation analysis? A: Yes, B-factors (temperature factors) are crucial for bridging static and dynamic views. High B-factor regions in a crystal structure often correspond to areas of high flexibility or disorder in simulations.
- Protocol for Comparative Analysis:
- Extract B-factors: From your PDB file, extract per-residue B-factors using a script (e.g., in Python/Biopython).
- Calculate RMSF from MD: From your MD trajectory, calculate the Root Mean Square Fluctuation (RMSF) for each Cα atom.
- Normalize and Correlate: Normalize both the B-factor and RMSF data (e.g., Z-score) and plot them on the same axis per residue. A strong positive correlation validates that your MD simulation recapitulates experimentally observed flexibility.
- Identify Discrepancies: Regions where simulation RMSF is low but experimental B-factors are high (or vice versa) are key. This may indicate over-constraining due to truncation or potential force field inaccuracies.
Q3: After running a long MD simulation for my designed enzyme, what specific metrics should I calculate to assess stability and function, particularly to diagnose over-truncation effects? A: Focus on metrics that probe structural integrity, flexibility, and functional geometry.
- Essential Analysis Protocol:
- Global Stability: Plot backbone RMSD over time. A stable, convergent trajectory is a prerequisite.
- Local Flexibility: Calculate per-residue RMSF. Map high-flexibility regions onto the 3D structure. Are they near the active site or truncation points?
- Active Site Integrity: Calculate the radius of gyration (Rg) for active site residues alone. Monitor distances between key catalytic residues. Significant drift suggests loss of functional architecture.
- Solvent Exposure & Interactions: Calculate the Solvent Accessible Surface Area (SASA) for the active site. Monitor the persistence of key hydrogen bonds or salt bridges lost due to truncation.
Q4: How can I use MD simulations to propose a correction for a protein model suspected of being over-truncated? A: MD can serve as a predictive tool for model correction.
- Iterative Refinement Protocol:
- Identify Dynamic "Breach" Points: From your RMSF and inter-residue distance analysis, identify regions where excessive flexibility may expose hydrophobic cores or disrupt networks.
- In-silico Mutagenesis & Loop Modeling: Use a tool like Rosetta or MODELLER to add back a limited number of residues N- or C-terminal to the breach point, or model a longer loop.
- Simulate the Corrected Model: Run a comparative MD simulation (50-100 ns) for both the original and corrected models under identical conditions.
- Quantitative Comparison: Compare the key metrics (see Table 1) between the two simulations. Improvement in stability (lower RMSD), recovery of key interactions, and correlation with experimental B-factors support the correction.
Table 1: Key Metrics for Diagnosing Over-Truncation Effects from MD Simulations
| Metric | Healthy/Full-Length System Indicator | Potential Over-Truncation Indicator | Calculation Tool/Code Example |
|---|---|---|---|
| Backbone RMSD | Plateaus at a low value (e.g., 1-3 Å). | Plateaus at a high value (>4-5 Å) or shows multiple sharp shifts. | gmx rms (GROMACS), cpptraj (AMBER) |
| Residue RMSF | Peaks in known flexible loops/tails; active site shows low fluctuation. | Exceptionally high peaks (>3 Å) at truncation sites or in core regions near the cut. | gmx rmsf, cpptraj |
| Active Site Rg | Remains relatively constant over time. | Shows a steady increase, indicating loss of compactness. | gmx gyrate (selecting active site residues) |
| Critical H-bond/Salt Bridge Occupancy | >80% occupancy throughout simulation. | <50% occupancy, indicating broken interaction network. | gmx hbond, VMD Hydrogen Bonds plugin |
| SASA of Active Site | Stable, correlating with substrate access channels. | Large, uncharacteristic increase, suggesting improper exposure or collapse. | gmx sasa |
Experimental Protocols
Protocol 1: Correlating Experimental B-factors with MD-derived RMSF
- Input: High-resolution (<2.5 Å) PDB file of the structure (wild-type or designed).
- B-factor Extraction: Use Biopython:
from Bio import PDB; parser=PDB.PDBParser(); structure=parser.get_structure('ID', 'file.pdb'); ... extract B-factor from atom objects. - MD Simulation: Perform a solvated, neutralized, energy-minimized, and equilibrated (NPT) production run of at least 100 ns.
- RMSF Calculation: After aligning the trajectory to the protein backbone, calculate RMSF for Cα atoms:
gmx rmsf -f traj.xtc -s topol.tpr -o rmsf.xvg -res - Correlation Analysis: Normalize both data sets (e.g.,
(value - mean)/std). Plot residue index vs. normalized B-factor and RMSF. Calculate Pearson correlation coefficient.
Protocol 2: Comparative Stability Analysis of Original vs. Corrected Model
- System Preparation: Prepare simulation systems for both the original truncated model and the corrected model using identical parameters (force field, water model, box size, ion concentration).
- Equilibration: Subject both systems to the exact same multi-step equilibration protocol (energy minimization, NVT, NPT).
- Production Runs: Run independent triplicate production simulations (3 x 100 ns) for each system. Use different random seeds for velocities.
- Convergence & Metrics Calculation: Discard the first 20 ns as equilibration. For each replicate, calculate all metrics in Table 1.
- Statistical Comparison: Perform a Student's t-test (or similar) on the averaged metric values (e.g., average RMSD, average active site Rg) from the triplicates of each system to determine if differences are statistically significant (p < 0.05).
Diagrams
Diagram 1: MD Analysis Workflow for Truncation Diagnosis
Diagram 2: Key Metric Relationship Network
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Comparative MD Analysis
| Item | Function in Analysis | Example/Note |
|---|---|---|
| High-Resolution Structural Data | Provides the initial atomic coordinates and experimental B-factors for validation. | PDB file from crystallography or cryo-EM. Aim for resolution <2.5 Å. |
| Molecular Dynamics Software | Engine to perform the physics-based simulations and generate trajectory data. | GROMACS, AMBER, NAMD, OpenMM. GROMACS is widely used for its speed. |
| Trajectory Analysis Suite | Tools to calculate quantitative metrics (RMSD, RMSF, Rg, SASA) from simulation trajectories. | Built-in tools in MD packages, MDTraj, MDAnalysis, VMD with TkConsole scripts. |
| Visualization Software | For 3D visualization of structures, trajectories, and mapping analysis results. | PyMOL, UCSF ChimeraX, VMD. Essential for intuitive understanding. |
| Scripting Environment | To automate analysis, correlate data, and generate custom plots. | Python (with NumPy, SciPy, Matplotlib, MDAnalysis), R, Bash scripting. |
| Comparative Model Builder | To generate corrected structural models by adding residues or remodeling loops. | MODELLER, RosettaCM, I-TASSER (for more extensive modeling). |
| Validation Server | To assess the geometric and stereochemical quality of initial and corrected models. | MolProbity, SAVES v6.0, PROCHECK. |
In Vitro vs. In Vivo Performance of Optimized Truncated Enzymes
Technical Support Center: Troubleshooting Enzyme Truncation Experiments
This support center is designed for researchers working within the thesis framework: "Addressing Over-Truncation in Enzyme Sequence Design: Bridging the In Silico, In Vitro, and In Vivo Efficacy Gap." The guides below address common pitfalls when translating optimized truncated enzymes from purified systems to complex biological environments.
FAQs & Troubleshooting
Q1: My truncated enzyme shows excellent catalytic efficiency (kcat/KM) in vitro but is completely inactive in cellular assays. What are the primary causes? A: This is a classic symptom of over-truncation. The likely causes are:
- Loss of Essential Post-Translational Modifications (PTMs): The truncated sequence may have removed sites for glycosylation, phosphorylation, or lipidation critical for in vivo stability, localization, or partner binding.
- Protein Misfolding & Aggregation: Truncation can remove critical structural elements or chaperone-binding sites, leading to misfolding and aggregation in the crowded cellular environment, despite proper folding in simplified in vitro conditions.
- Increased Proteolytic Degradation: The removal of domains that confer structural stability or protective motifs (e.g., PAS domains) can expose protease cleavage sites, leading to rapid intracellular degradation.
- Subcellular Mislocalization: Deletion of signaling peptides (e.g., nuclear localization signals, membrane anchors) prevents the enzyme from reaching its correct subcellular compartment.
Q2: How can I systematically diagnose the cause of poor in vivo performance? A: Follow this diagnostic workflow to isolate the failure point.
Diagram Title: Diagnostic Workflow for In Vivo Enzyme Failure
Q3: What are the key quantitative benchmarks for comparing in vitro vs. in vivo performance? A: Monitor these parameters in parallel. A significant drop in the In Vivo:In Vitro Performance Ratio indicates a context-specific failure.
Table 1: Key Performance Indicators (KPIs) for Truncated Enzymes
| Parameter | In Vitro Measurement | In Vivo Measurement | Acceptable Discrepancy Threshold (Thesis Guideline) |
|---|---|---|---|
| Catalytic Efficiency | kcat/KM (purified enzyme) | Rate of product formation in cell lysate (normalized to expression) | ≤ 10-fold reduction |
| Protein Stability | Tm (thermal melt), t1/2 (accelerated degradation) | Cellular half-life (cycloheximide chase) | ≥ 60% of full-length half-life |
| Effective Concentration | Active site titration | Functional expression level (quantitative WB / activity) | ≥ 30% of in vitro assay concentration |
| Specific Activity | μmol product/min/mg (pure) | μmol product/min/mg (from lysate IP) | ≤ 5-fold reduction |
Q4: My truncated enzyme is stable but inactive in vivo. Could cofactor or effector binding be affected? A: Yes. Truncation often removes allosteric or structural domains that modulate cofactor affinity (e.g., NADPH, metal ions) which may be present at saturating levels in vitro but at limiting concentrations in vivo. Perform a Cofactor Rescue Experiment.
Protocol: Cofactor/Effector Rescue Assay
- Transfert cells with your truncated enzyme construct.
- Treat cells with cell-permeable forms of the suspected cofactor (e.g., MgCl₂, ZnCl₂, methyl-NAD) or a known allosteric effector at physiological doses.
- Lyse cells after 24-48 hours treatment.
- Measure Activity in clarified lysates using a specific, sensitive fluorogenic or chromogenic substrate. Normalize activity to enzyme expression level via Western blot.
- Compare to full-length enzyme and untreated truncated controls. Restoration of activity points to a cofactor affinity issue caused by truncation.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Evaluating Truncated Enzymes
| Reagent / Material | Function in Experiment | Critical Note for Truncation Studies |
|---|---|---|
| Thermofluor Dyes (e.g., SYPRO Orange) | Measure protein thermal stability (Tm) in vitro. | Identifies destabilization from truncation. A ΔTm > 5°C vs. full-length is a red flag. |
| Proteasome Inhibitor (e.g., MG132) | Inhibits the ubiquitin-proteasome system. | If truncated enzyme activity/stability improves in cells with MG132, it confirms targeted degradation. |
| Cell-Permeable Cofactors | Rescue experiments for metal-dependent or cofactor-requiring enzymes. | Diagnoses if truncation lowered cofactor affinity to non-physiological levels. |
| FRET-based Folding Biosensor | Reports on intracellular protein folding state. | Directly tests if truncation causes misfolding in the cellular environment. |
| Cross-linking Agent (e.g., DSS) | Stabilizes weak protein-protein interactions. | Can capture transient interactions with chaperones or protective partners lost due to truncation. |
| Subcellular Fractionation Kit | Isolates organelles (cytosol, membranes, nuclei). | Confirms correct localization; mislocalization is a common truncation artifact. |
| Live-Cell, Membrane-Permeant Enzyme Substrate | Measures real-time activity in living cells (e.g., fluorogenic esterase probes). | Provides the most direct in vivo activity readout, independent of lysis artifacts. |
Q5: Based on current research, what is a failsafe step before finalizing a truncation design? A: Conduct a Minimal Functional Domain Validation Cascade in silico and in vitro before moving to cellular models.
Diagram Title: Validation Cascade to Prevent Over-Truncation
Thesis-Context Summary: The core challenge is that in vitro optimization often selects for the minimal catalytic core, while in vivo functionality requires a minimal functional unit that includes elements for stability, localization, and regulation. The troubleshooting guides above aim to diagnose and rectify the removal of these critical, context-dependent elements, thereby addressing the central thesis problem of over-truncation.
Technical Support Center: Troubleshooting Guide & FAQs
Question: What are the primary experimental indicators of a successful versus a failed truncation? Answer: A successful truncation maintains or enhances catalytic efficiency (kcat/KM) and stability (Tm, t1/2), while a failed truncation shows significant losses in these parameters. Key metrics for comparison are summarized below.
Question: My truncated construct shows poor solubility. What are the first steps to troubleshoot? Answer: Poor solubility often indicates exposed hydrophobic cores or loss of stabilizing interactions. First, verify the truncation boundaries via homology modeling to ensure you haven't removed critical secondary structure elements. Perform a thermal shift assay to check for major destabilization. Consider adding a solubility tag for expression and testing refolding protocols.
Question: How can I predict if a truncation will disrupt allosteric or regulatory sites? Answer: Analyze conserved sequence motifs and known functional domains from databases like Pfam and InterPro prior to design. Use tools like ConSurf to map evolutionary conservation. Experimentally, compare the kinetics of the truncated enzyme with the full-length enzyme in the presence and absence of known allosteric modulators.
Question: The truncated enzyme expresses well but is inactive. What could be the cause? Answer: This typically indicates improper folding or removal of a critical catalytic residue or loop. Check the following:
- Folding: Use circular dichroism (CD) spectroscopy to compare secondary structure with the wild-type.
- Active Site Integrity: Perform a binding assay with a substrate analog or inhibitor.
- Critical Loops: Review literature and structural data (e.g., Catalytic Site Atlas) to ensure no essential flexible loops were deleted.
Experimental Protocols
Protocol 1: In Silico Stability Prediction for Truncation Design Method: Use RosettaDDGPrediction or FoldX to calculate the change in free energy of folding (ΔΔG) for the truncated structure compared to the full-length enzyme. Model the truncation using a high-quality parent structure (PDB). Run 50 iterations per mutant and average the results. A ΔΔG > 2-3 kcal/mol often predicts significant destabilization.
Protocol 2: High-Throughput Screening of Truncation Libraries for Solubility and Stability Method: Clone truncation variants into a vector with a C-terminal GFP fusion. Express in E. coli in a 96-well format. Measure culture fluorescence (ex488/em510) after induction as a proxy for soluble fusion protein. In parallel, lyse cells and measure catalytic activity with a fluorescent substrate. Normalize activity to soluble GFP signal to identify variants with high specific activity.
Protocol 3: Determining Thermostability (Tm) via Differential Scanning Fluorimetry (DSF) Method: Purify wild-type and truncated enzymes. Set up reactions in a real-time PCR machine: 5 µM enzyme, 5X SYPRO Orange dye, in assay buffer (e.g., 20 mM HEPES, 150 mM NaCl, pH 7.5). Perform a temperature ramp from 25°C to 95°C at 1°C/min, monitoring fluorescence. Plot the derivative of fluorescence (RFU) over temperature to determine the Tm (melting temperature).
Data Presentation
Table 1: Comparative Metrics of Successful vs. Failed Truncations in Clinical Candidate Enzymes
| Metric | Successful Truncation (e.g., HCV NS3 Protease ΔC-term) | Failed Truncation (e.g., Truncated PTP1B ΔIRS1) | Measurement Method |
|---|---|---|---|
| Catalytic Efficiency (kcat/KM) | ≥ 90% of wild-type | < 20% of wild-type | Michaelis-Menten kinetics |
| Thermal Stability (Tm) | ΔTm ≤ -2°C | ΔTm ≤ -10°C | Differential Scanning Fluorimetry (DSF) |
| Half-life (t1/2, 37°C) | ≥ 80% of wild-type | ≤ 30% of wild-type | Activity decay over time |
| Solubility (Yield) | > 10 mg/L from E. coli | < 1 mg/L, forms inclusion bodies | Purification yield (A280) |
| Aggregation Propensity | Low (monomeric by SEC) | High (multimeric/aggregated by SEC) | Size-Exclusion Chromatography |
| Structural Integrity (RMSD) | < 2.0 Å backbone deviation | > 4.0 Å backbone deviation | X-ray Crystallography/NMR |
Table 2: Research Reagent Solutions Toolkit
| Reagent / Material | Function in Truncation Analysis |
|---|---|
| SYPRO Orange Dye | Binds hydrophobic patches exposed during thermal denaturation in DSF assays. |
| Size-Exclusion Chromatography (SEC) Standards | Calibrates column to determine oligomeric state and aggregation of purified truncations. |
| Protease Inhibitor Cocktail (e.g., EDTA-free) | Prevents unintended proteolysis of unstable truncated enzymes during purification. |
| His-tag Purification Resin (Ni-NTA/Co2+) | Enables rapid immobilization and purification of tagged truncation constructs for analysis. |
| Thermostable Polymerase for SDM | Used for site-directed mutagenesis to create precise truncation boundaries. |
| Circular Dichroism (CD) Buffer Kit | Provides optimized, UV-transparent buffers for accurate secondary structure assessment. |
| FRET-based Substrate Analogue | Allows sensitive, continuous activity measurement for kinetic profiling of truncations. |
Mandatory Visualizations
Title: Successful Enzyme Truncation Design Workflow
Title: Primary Pathways Leading to Failed Enzyme Truncation
Establishing Best-Practice Validation Frameworks for the Field
Technical Support Center
FAQ & Troubleshooting Guide
Q1: My designed enzyme shows strong in vitro activity but fails completely in the cellular assay. What could be the cause?
- A: This is a classic symptom of over-truncation. Your design may have removed regulatory domains or intrinsic disorder regions essential for in vivo stability, localization, or participation in protein-protein interaction networks. Recommended Action: Perform a co-evolution analysis (e.g., using DeepMutualEvo) on the full-length native sequence to identify potentially critical long-range interactions. Re-design to include identified key regions or develop a fusion construct.
Q2: How do I determine if my sequence has been over-truncated?
- A: Implement the following diagnostic checklist:
- Compare to Natural Variants: Use Pfam and InterPro to see if your designed construct omits entire domains present in all natural orthologs.
- Predict Disordered Regions: Run predictors like IUPred2A or AlphaFold3. A good design often retains critical short disordered linkers.
- Stability Check: Use Rosetta
ddg_monomeror FoldX to calculate ΔΔG. An over-stable (excessively negative ΔΔG) protein may be rigid and non-functional. - Aggregation Propensity: Check with tools like Aggrescan. Exposed hydrophobic cores from truncation can increase aggregation risk.
- A: Implement the following diagnostic checklist:
Q3: I am getting high expression yields, but my protein is largely insoluble. How can I troubleshoot this?
- A: Insolubility following truncation often points to improper folding due to missing structural elements.
- Step 1: Analyze the solubility of your construct with SoluProt.
- Step 2: Run AGGRESCAN on the full-length sequence to see if your truncation accidentally exposed a hydrophobic aggregation "hot spot."
- Step 3: Consider adding solubility tags (e.g., MBP, SUMO) or designing conservative point mutations to increase surface polarity, rather than deleting large segments.
- A: Insolubility following truncation often points to improper folding due to missing structural elements.
Q4: What are the key metrics to include in a validation framework to prevent over-truncation artifacts?
- A: A robust framework should include orthogonal metrics beyond mere expression and basic activity. See the table below.
Table 1: Core Validation Metrics for Truncation Designs
| Validation Tier | Metric | Target Value/Range | Purpose in Addressing Over-Truncation |
|---|---|---|---|
| Biophysical | Thermostability (Tm) | ΔTm vs. baseline < ±5°C | Large increases may indicate unnatural rigidity; decreases indicate destabilization. |
| Polydispersity (DLS/SEC-MALS) | PDI < 0.2; Monodisperse peak | Ensures homogeneity; high PDI suggests aggregation or misfolding. | |
| Melting Curve Width (nanoDSF) | FWHM < 8°C | Broad transitions suggest heterogeneous or non-cooperative folding. | |
| Functional | Catalytic Efficiency (kcat/Km) | > 10% of native full-length | Confirms active site integrity is not perturbed. |
| Specific Activity in Cell Lysate | Correlation > 0.8 with purified | Tests performance in complex, in vivo-like environment. | |
| In-cell | Protein Turnover (Half-life) | > 50% of full-length control | Ensures truncation did not create a degradation signal. |
| Correct Subcellular Localization | Match to expected pattern | Confirms retention of targeting sequences. |
Experimental Protocols
Protocol 1: Integrated In Vitro to In Vivo Activity Correlation Assay
- Objective: To directly identify discrepancies between purified enzyme performance and cellular function.
- Materials: See "The Scientist's Toolkit" below.
- Method:
- Express & Purify: Express your truncated design and a full-length control in E. coli (or relevant host). Purify using His-tag affinity chromatography.
- In Vitro Assay: Measure kinetic parameters (kcat, Km) for both constructs under identical, physiologically relevant buffer conditions.
- In Vivo Assay: In parallel, transform both constructs into your target cellular system (e.g., yeast, mammalian cells). Prepare whole-cell lysates via gentle lysis.
- Normalization: Quantify protein expression in lysates via Western blot or quantitative fluorescence. Normalize activity measurements in lysates to protein concentration.
- Correlation Analysis: Plot in vitro specific activity vs. in-cell normalized activity. A significant outlier trend for truncation designs indicates a context-dependent functional loss.
Protocol 2: Differential Scanning Fluorimetry (nanoDSF) for Folding Cooperativity
- Objective: To assess the folding "tightness" and homogeneity of a truncated design.
- Method:
- Prepare protein samples at 0.5 mg/mL in assay buffer.
- Load samples into nanoDSF-grade glass capillaries.
- Use a Prometheus NT.48 or similar system to apply a thermal ramping profile (1°C/min from 20°C to 95°C).
- Monitor intrinsic tryptophan fluorescence at 330 nm and 350 nm simultaneously.
- Analyze the first derivative of the 350nm/330nm ratio to determine the melting temperature (Tm) and the Full Width at Half Maximum (FWHM) of the transition peak. A broad FWHM suggests non-two-state unfolding, indicative of a misfolded or heterogeneous population.
Visualizations
Title: Framework for Validated Truncation Design
Title: Over-Truncation Causes and Observed Symptoms
The Scientist's Toolkit: Key Research Reagent Solutions
| Item | Function in Validation |
|---|---|
| HisTrap HP Column (Cytiva) | Standardized affinity chromatography for high-yield, high-purity protein purification of tagged constructs. |
| Prometheus NT.48 (NanoTemper) | nanoDSF system for label-free measurement of protein thermal stability and folding cooperativity. |
| Superdex 200 Increase (Cytiva) | Size-exclusion chromatography column for assessing aggregation state and monodispersity (SEC). |
| SpectraMax M5e (Molecular Devices) | Multi-mode microplate reader for high-throughput kinetic assays and fluorescence-based activity screens. |
| Proteostat Aggregation Assay (Enzo) | Dye-based fluorescence assay to quantify protein aggregation in solution or in cells. |
| Anti-Strep-tag II Antibody (HRP-conj.) | For uniform Western blot detection and quantification of Strep-tagged constructs across designs. |
| HaloTag Mammalian Expression System (Promega) | Enables covalent labeling for precise in-cell localization and turnover studies. |
| Sf9 Insect Cells & Baculovirus System | Eukaryotic expression system for producing complex, multi-domain proteins requiring post-translational modifications. |
Conclusion
Effectively addressing over-truncation requires a balanced, data-driven approach that prioritizes functional integrity over mere sequence minimization. By understanding its foundational causes, implementing preventive design methodologies, applying robust troubleshooting protocols, and adhering to rigorous comparative validation, researchers can design truncated enzymes that are both stable and catalytically potent. Future directions involve the integration of generative AI and high-throughput functional screening to create ultra-stable minimal enzymes, directly impacting the development of more efficacious, manufacturable, and deliverable enzyme therapeutics for a wide range of biomedical applications, from metabolic disorders to targeted prodrug activation.