De Novo Enzyme Design: Engineering Novel Biocatalysts from Scratch for Biomedical Applications
This article provides a comprehensive overview of the rapidly advancing field of de novo enzyme design, a discipline that creates novel protein catalysts with functions not found in nature.
De Novo Enzyme Design: Engineering Novel Biocatalysts from Scratch for Biomedical Applications
Abstract
This article provides a comprehensive overview of the rapidly advancing field of de novo enzyme design, a discipline that creates novel protein catalysts with functions not found in nature. Tailored for researchers, scientists, and drug development professionals, we explore the foundational principles driving the need for artificial enzymes, from overcoming the limitations of natural biocatalysts to enabling abiotic reactions like olefin metathesis in living systems. The content delves into the integrated methodological toolkit, combining computational design, artificial intelligence, and directed evolution. It further addresses critical challenges in optimization and troubleshooting, and concludes with a rigorous examination of validation frameworks and comparative analyses of current technologies, highlighting their profound implications for therapeutic development and green chemistry.
Beyond Nature's Blueprint: The Rationale and Core Principles of Designing Enzymes from Scratch
The Catalytic Limitations of Natural Enzymes and the Case for De Novo Design
Natural enzymes, often described as "nature's privileged catalysts," are renowned for their exceptional selectivity and efficiency in accelerating biochemical reactions under mild physiological conditions [1] [2]. These protein catalysts drive virtually all essential biological processes, from cell growth and development to complex material synthesis in living organisms [2]. However, despite their evolutionary optimization for specific biological functions, natural enzymes possess inherent limitations that restrict their utility in industrial, pharmaceutical, and research contexts. These constraints include narrow substrate specificity, instability under non-physiological conditions, and an inability to catalyze "new-to-nature" reactions not found in biological systems [1] [3].
The field of enzyme engineering has emerged to overcome these limitations, with de novo enzyme design representing a paradigm shift from modifying existing enzymes to creating entirely new catalytic proteins from first principles [3]. This approach uses computational and artificial intelligence (AI) methodologies to design novel enzyme sequences and structures tailored for specific applications, bypassing the constraints of natural enzyme evolution [4] [3]. By moving beyond the limitations of natural enzyme scaffolds, de novo design promises to unlock new possibilities in sustainable chemistry, drug development, and biotechnology through the creation of biocatalysts with customized functions, enhanced stability, and novel catalytic mechanisms [1] [2] [3].
Fundamental Limitations of Natural Enzyme Catalysis
Restricted Reaction Scope and Functional Limitations
Natural enzymes have evolved to catalyze a specific set of biochemical reactions essential for life processes, creating significant gaps in their catalytic repertoire for synthetic chemistry applications. This limitation becomes particularly evident in several key areas:
- Lack of efficient natural enzymes for synthetically valuable bond-forming reactions, including carbon-carbon and carbon-silicon bond formation, which are fundamental to pharmaceutical and materials synthesis [1]
- Inability to catalyze abiotic reactions not found in nature, such as the Kemp elimination, Diels-Alder cycloadditions, or olefin metathesis, despite the potential utility of these transformations in synthetic chemistry [5] [6]
- Limited capacity for industrial substrate processing, as natural enzymes typically recognize only specific biological molecules under narrow environmental conditions [1]
Operational Instability Under Non-Physiological Conditions
Natural enzymes function within precise physiological environments, but industrial and pharmaceutical applications often require stability under drastically different conditions:
- Narrow functional ranges for parameters such as temperature, pH, and ionic strength, outside of which enzymatic activity rapidly declines [1] [7]
- Susceptibility to organic solvents used in industrial processes, with most natural enzymes denaturing in solvent concentrations above 10-20% [1]
- Thermolability at elevated temperatures used in industrial processes to increase reaction rates and reduce microbial contamination [8]
- Proteolytic degradation in therapeutic applications, limiting their half-life and efficacy in biological systems [8]
Structural Inefficiencies and Delivery Challenges
The complex architecture of natural enzymes presents practical challenges for various applications:
- Large size and structural redundancy, where much of the protein scaffold may contribute minimally to catalytic function while increasing metabolic costs of synthesis and folding [8]
- Delivery limitations in therapeutic applications, where large enzymes like CRISPR-Cas systems face challenges in efficient delivery to target tissues and cells due to vector payload constraints [8]
- Reduced electron transfer rates in biosensing applications, where increased cofactor-electrode distances in large oxidoreductases compromise detection sensitivity [8]
Table 1: Key Limitations of Natural Enzymes in Industrial and Therapeutic Applications
| Limitation Category | Specific Constraints | Impact on Applications |
|---|---|---|
| Functional Scope | Limited to biological reactions; Cannot catalyze abiotic transformations | Restricts use in synthetic chemistry and new reaction development |
| Environmental Sensitivity | Narrow pH and temperature ranges; Organic solvent intolerance | Limits process conditions in manufacturing; Requires costly stabilization |
| Structural Issues | Large size with redundant elements; Flexible loops and disordered regions | Challenges in therapeutic delivery; Reduced thermal stability; Folding inefficiencies |
| Catalytic Efficiency | Optimized for physiological substrates; Product inhibition issues | Poor performance with non-natural substrates; Low productivity in industrial processes |
The Emergence of De Novo Enzyme Design
Fundamental Principles and Definitions
De novo enzyme design represents a computational approach to creating novel protein sequences and structures from first principles or learned models, rather than modifying existing natural enzymes [3]. This methodology stands in contrast to traditional enzyme engineering approaches such as rational design (targeted mutations based on structure) and directed evolution (iterative rounds of mutation and selection) [2] [3]. The core premise of de novo design is the ability to access entirely novel regions of sequence-structure space, unconstrained by the evolutionary history of natural enzymes [3].
This approach typically begins with a theozyme (theoretical enzyme) - a computational model of the ideal catalytic constellation for a target reaction, often derived from quantum-mechanical calculations of the transition state [4] [6]. Design methods then identify or generate protein scaffolds capable of positioning amino acid side chains to precisely stabilize this transition state, creating an environment optimal for catalysis [6]. The designs are subsequently refined using physics-based energy functions and, increasingly, artificial intelligence methods to ensure stable folding and catalytic competence [4] [3].
Comparative Analysis: Traditional Engineering vs. De Novo Approaches
Table 2: Comparison of Enzyme Engineering Methodologies
| Methodology | Key Principles | Advantages | Limitations |
|---|---|---|---|
| Rational Design | Structure-based mutagenesis of natural enzymes; Focus on active site engineering | High precision; Minimal mutations required; Clear structure-function relationships | Limited by natural scaffold constraints; Requires extensive structural knowledge |
| Directed Evolution | Iterative rounds of random mutagenesis and screening/selection for desired traits | No structural knowledge needed; Can discover unexpected solutions; Proven industrial track record | Labor-intensive; Limited exploration of sequence space; Local optimization |
| De Novo Design | Computational creation from first principles; Transition state stabilization in novel scaffolds | Access to novel folds and functions; Not limited by natural enzyme repertoire; Global sequence space exploration | Computational complexity; Challenges in predicting folding and function; Often requires experimental refinement |
Methodological Framework for De Novo Enzyme Design
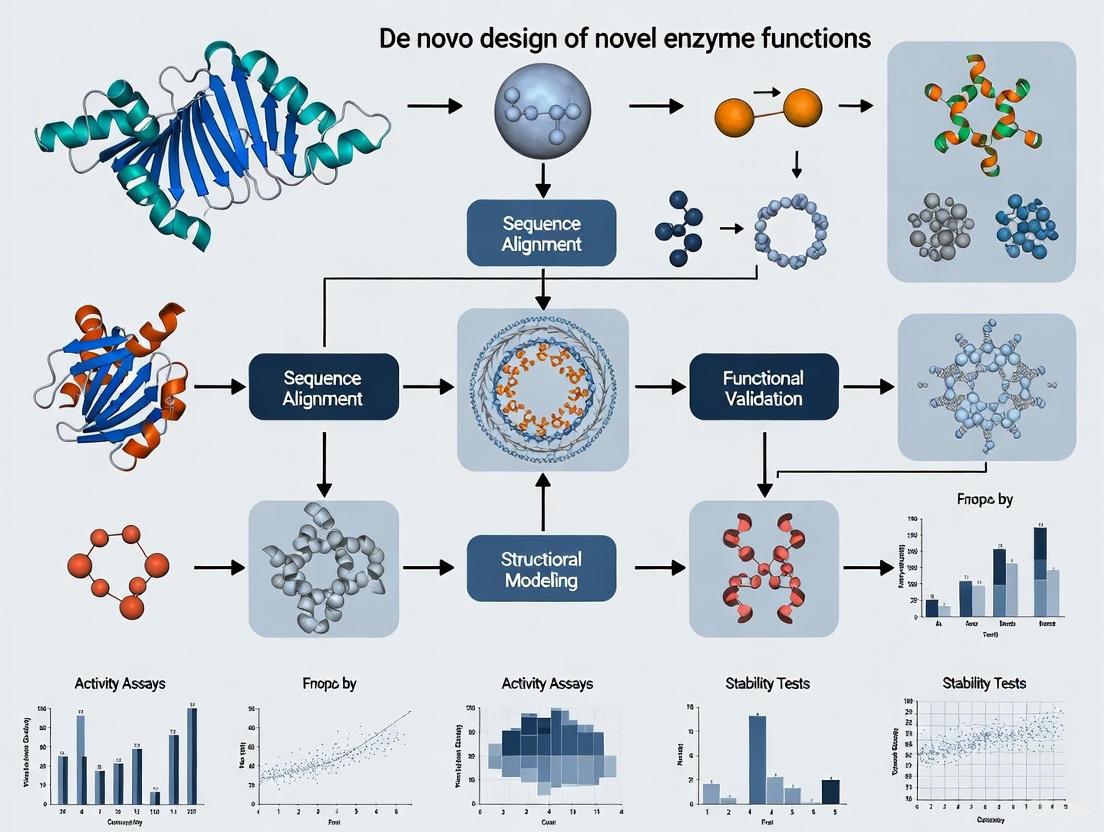
Computational Workflows and Design Strategies
The de novo design process follows structured computational workflows that integrate multiple methodologies to create functional enzymes. Recent advances have demonstrated complete computational workflows that generate efficient enzymes without requiring extensive experimental optimization [6]. The core strategies include:
- Structure-based design: Utilizes physical energy functions and spatial pattern algorithms to derive stable conformations from 3D structural constraints, often employing frameworks like helical bundle proteins or TIM-barrel folds [3] [6]
- Sequence-based design: Employs deep generative models trained on natural protein sequences to learn co-evolutionary patterns and generate functional sequences from data-driven principles [3]
- Fragment-based backbone generation: Combines structural fragments from natural proteins to create novel backbones with diverse active-site geometries [6]
- Inside-out design: Models the catalytic transition state first, then builds the supporting protein scaffold around this functional core [4]
The following diagram illustrates a representative workflow for computational de novo enzyme design:
Key Experimental Protocols and Methodologies
De Novo Design of Kemp Eliminases
The Kemp elimination reaction serves as a benchmark for de novo enzyme design, as no natural enzyme is known to catalyze this proton transfer from carbon [6]. A recently published protocol demonstrates a fully computational workflow for designing high-efficiency Kemp eliminases:
Theozyme Construction: Quantum mechanical calculations define the ideal catalytic constellation, including a carboxylate base (Asp/Glu) for proton abstraction and aromatic residues for π-stacking with the substrate transition state [6]
Backbone Generation: Thousands of TIM-barfold backbones are generated using combinatorial assembly of fragments from natural proteins (e.g., indole-3-glycerol-phosphate synthase family) to create structural diversity in active site regions [6]
Transition State Placement: Geometric matching algorithms position the KE theozyme in each generated backbone, identifying scaffolds with optimal preorganization for catalysis [6]
Active Site Optimization: Rosetta atomistic calculations mutate all active-site positions to optimize interactions with the transition state while maintaining low energy states [6]
Filtering and Selection: A "fuzzy-logic" optimization objective function balances conflicting design criteria (catalytic geometry, desolvation of catalytic base, and overall protein stability) to select top designs [6]
Stability Enhancement: Comprehensive stabilization of the active site and protein core through sequence optimization, often resulting in designs with >100 mutations from any natural protein [6]
This protocol has yielded Kemp eliminases with catalytic efficiencies of 12,700 M⁻¹s⁻¹ and rates of 2.8 s⁻¹, surpassing previous computational designs by two orders of magnitude and approaching the efficiency of natural enzymes [6].
Artificial Metathase Design for Olefin Metathesis
Olefin metathesis represents a powerful carbon-carbon bond formation reaction with no natural enzyme equivalent. The design protocol for artificial metathases involves:
Cofactor Design: Engineering a Hoveyda-Grubbs catalyst derivative (Ru1) with polar sulfamide groups to enable supramolecular interactions with the protein scaffold and improve aqueous solubility [5]
Scaffold Selection: Using de novo-designed closed alpha-helical toroidal repeat proteins (dnTRP) as hyperstable scaffolds with engineerable binding pockets [5]
Computational Docking: Using RifGen/RifDock suites to enumerate amino acid rotamers around the cofactor and dock the ligand with interacting residues into protein cavities [5]
Binding Affinity Optimization: Iterative design of hydrophobic contacts (e.g., Phe→Trp mutations at positions F43 and F116) to enhance cofactor binding (KD ≤ 0.2 μM) through supramolecular anchoring [5]
Directed Evolution: Engineering catalytic performance through screening in E. coli cell-free extracts at optimized pH (4.2) with glutathione oxidation using Cu(Gly)₂ [5]
This approach has produced artificial metathases with turnover numbers ≥1,000 for ring-closing metathesis in cytoplasmic environments, demonstrating pronounced biocompatibility and catalytic efficiency [5].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagents and Computational Tools for De Novo Enzyme Design
| Tool Category | Specific Tools/Reagents | Function/Application |
|---|---|---|
| Computational Design Suites | Rosetta Macromolecular Modeling Suite; RifGen/RifDock | Protein structure prediction, design, and ligand docking; Energy-based scoring and optimization |
| AI/ML Platforms | ProteinMPNN; AlphaFold 2/3; ESMFold; Generative Language Models | Protein sequence design; Structure prediction from sequence; Generation of novel protein sequences |
| Expression Systems | E. coli BL21(DE3); Pichia pastoris; Cell-free expression systems | Heterologous protein expression; Rapid screening of design variants |
| Characterization Methods | X-ray crystallography; Native mass spectrometry; Tryptophan fluorescence quenching | Structural validation; Binding affinity measurements (KD); Complex stoichiometry determination |
| Activity Assays | UV-Vis spectroscopy (Kemp elimination); GC-MS (metathesis products); HPLC analysis | Kinetic parameter determination (kcat, KM); Reaction monitoring and product identification |
| Stability Assessment | Differential scanning calorimetry; Circular dichroism; Thermal shift assays | Melting temperature (Tm) determination; Secondary structure analysis; Stability profiling |
Case Studies and Experimental Validation
Breakthrough Designs and Catalytic Performance
Recent advances in de novo enzyme design have yielded several breakthrough catalysts that demonstrate the field's rapid progress:
High-Efficiency Kemp Eliminases: A fully computational design workflow has produced Kemp eliminases with remarkable catalytic parameters, including kcat/KM = 12,700 M⁻¹s⁻¹ and kcat = 2.8 s⁻¹ [6]. These designs featured more than 140 mutations from any natural protein and exhibited exceptional thermal stability (>85°C). Further optimization through computational active-site redesign achieved catalytic efficiencies exceeding 10⁵ M⁻¹s⁻¹ with turnover rates of 30 s⁻¹, matching the performance of natural enzymes and challenging fundamental assumptions about biocatalytic design limitations [6].
Artificial Metathases for Whole-Cell Biocatalysis: De novo-designed artificial metalloenzymes incorporating synthetic ruthenium cofactors have enabled olefin metathesis—a reaction with no natural biological counterpart—within living E. coli cells [5]. These designs combined tailored Hoveyda-Grubbs catalyst derivatives with hyperstable de novo protein scaffolds, achieving turnover numbers ≥1,000 for ring-closing metathesis of olefins in cytoplasmic environments. The integration of computational design with directed evolution resulted in variants with excellent catalytic performance and pronounced biocompatibility [5].
Carbon-Silicon Bond Formation: Researchers have developed a workflow converting simple miniature helical bundle proteins into efficient and selective enzymes for forming carbon-silicon bonds, addressing a significant gap in natural enzymatic capabilities [1]. This approach combined de novo protein design with state-of-the-art artificial intelligence methods to create sequences that support non-biological transformations, demonstrating the potential for creating enzymes that operate via mechanisms not previously known in nature [1].
Comparative Performance Analysis
Table 4: Catalytic Performance of Representative De Novo Designed Enzymes
| Enzyme Design | Reaction Type | Catalytic Efficiency (kcat/KM) | Turnover Number (kcat) | Thermal Stability |
|---|---|---|---|---|
| Kemp Eliminase Des27 [6] | Proton transfer | 12,700 M⁻¹s⁻¹ | 2.8 s⁻¹ | >85°C |
| Optimized Kemp Eliminase [6] | Proton transfer | >100,000 M⁻¹s⁻¹ | 30 s⁻¹ | >85°C |
| Artificial Metathase dnTRP_18 [5] | Olefin metathesis | N/R | ≥1,000 | T₅₀ >98°C |
| Previous KE Designs [6] | Proton transfer | 1-420 M⁻¹s⁻¹ | 0.006-0.7 s⁻¹ | Variable |
| Natural Enzymes (median) [6] | Various | ~100,000 M⁻¹s⁻¹ | ~10 s⁻¹ | Variable |
N/R: Not reported in the source material
Advantages and Applications of De Novo Designed Enzymes
Functional and Operational Benefits
De novo designed enzymes offer significant advantages over both natural enzymes and traditional engineered variants:
- Expanded reaction scope: Capacity to catalyze non-biological transformations including carbon-silicon bond formation, olefin metathesis, and Kemp elimination [1] [5] [6]
- Enhanced stability characteristics: Hyperstable designs with T₅₀ >98°C and tolerance to extreme pH conditions (2.6-8.0), far exceeding typical natural enzyme stability ranges [5]
- Organic solvent compatibility: Function in up to 60% organic solvent concentrations, enabling chemistry in environments incompatible with natural enzymes [1]
- Customizable cofactor utilization: Ability to incorporate non-biological cofactors and metal complexes for abiotic catalysis [1] [5]
- Size optimization: Miniature enzyme designs with improved folding efficiency, expressibility, thermostability, and resistance to proteolysis [8]
Industrial and Therapeutic Applications
The unique properties of de novo designed enzymes open new possibilities across multiple domains:
Pharmaceutical Manufacturing:
- Synthesis of complex active pharmaceutical ingredients (APIs) through biocatalytic routes with improved selectivity and reduced environmental impact [9]
- Enzyme-based therapies for genetic disorders (e.g., acute lymphoblastic leukemia with asparaginase) through engineered specificity and reduced immunogenicity [9]
- Targeted cancer treatments using enzymes designed to selectively activate prodrugs within tumor environments [10] [9]
Sustainable Chemistry:
- Biocatalysts for green chemistry applications operating in water as the "greenest solvent" with reduced energy requirements [1]
- Degradation of environmental pollutants through enzymes with tailored activities for emerging contaminants [2]
- Biosensing platforms with miniature enzymes exhibiting enhanced electron transfer rates for improved detection sensitivity [8]
Advanced Materials and Synthesis:
- Custom catalysts for polymer synthesis and materials design through controlled bond-forming reactions [1]
- Industrial biocatalysis under demanding process conditions incompatible with natural enzymes [3] [8]
Future Directions and Concluding Perspectives
Emerging Trends and Methodological Advances
The field of de novo enzyme design is rapidly evolving, driven by several converging technological developments:
- Artificial Intelligence Integration: Machine learning models, particularly deep generative networks and protein language models, are revolutionizing sequence-structure-function predictions, enabling more accurate design of functional proteins [4] [2] [3]
- Automated Design Workflows: Movement toward general, automated protein design systems that unify sequence generation, structure prediction, and fitness optimization in integrated frameworks [2]
- High-Throughput Characterization: Development of rapid experimental validation methods to generate labeled training data for AI models, creating virtuous cycles of design improvement [2]
- Miniaturization Strategies: Increased focus on designing compact enzymes with improved folding kinetics, stability, and delivery characteristics for therapeutic and diagnostic applications [8]
De novo enzyme design represents a fundamental shift in our approach to creating biological catalysts, moving beyond the constraints of natural enzyme evolution to rationally design proteins with tailored functions. While natural enzymes will continue to serve important roles in biocatalysis, their inherent limitations in reaction scope, stability, and customizability create compelling opportunities for designed alternatives.
The recent success in creating highly efficient enzymes through completely computational workflows [6], combined with the ability to catalyze abiotic reactions in biological environments [5], demonstrates that de novo design has transitioned from theoretical possibility to practical capability. As AI methodologies continue to advance and our understanding of protein folding and catalysis deepens, we can anticipate increasingly sophisticated designs that further blur the distinction between natural and artificial enzymes.
For researchers and drug development professionals, these developments offer unprecedented opportunities to create custom biocatalytic solutions for specific challenges, from sustainable chemical synthesis to targeted therapeutic interventions. The coming years will likely see de novo designed enzymes moving from laboratory demonstrations to broad industrial and clinical application, ultimately fulfilling the promise of tailor-made catalysts designed from first principles.
Defining Artificial Metalloenzymes (ArMs) and New-to-Nature Reactions
Artificial metalloenzymes (ArMs) represent a pioneering class of designer biocatalysts that combine the versatile reactivity of synthetic metallocatalysts with the precise selectivity of protein scaffolds. These hybrid catalysts are not found in nature and are engineered to catalyze both natural reactions with enhanced selectivity and new-to-nature reactions—chemical transformations without precedent in biological systems [11]. The fundamental architecture of an ArM consists of two primary components: a genetically engineerable protein scaffold that provides a defined second coordination sphere, and an artificial catalytic moiety featuring a synthetic metal center that enables novel reactivity [11] [12].
The significance of ArMs extends across multiple disciplines, from synthetic chemistry to pharmaceutical development. They effectively bridge the gap between homogeneous catalysis and enzymatic catalysis, offering the potential to perform reactions in water under mild conditions while maintaining the high activity and broad reaction scope typical of organometallic catalysts [12]. This unique combination addresses longstanding challenges in synthetic chemistry, including the catalytic asymmetric synthesis of complex molecules and the implementation of sustainable chemical processes.
Structural Composition and Assembly Strategies
Fundamental Components
The performance of ArMs derives from the synergistic interaction between their constituent parts. The metal cofactor provides the primary catalytic activity, often enabling reaction mechanisms inaccessible to purely biological systems. These cofactors can range from simple metal ions to sophisticated organometallic complexes. The protein scaffold serves multiple critical functions: it creates a chiral environment to enforce enantioselectivity, enhances catalyst stability through encapsulation, and provides a platform for iterative optimization through protein engineering [11].
The development of ArMs has been accelerated through chemogenetic optimization, a parallel improvement strategy that simultaneously refines both the direct metal surroundings (first coordination sphere) and the protein environment (second coordination sphere) [11]. This approach allows researchers to fine-tune catalytic properties through a combination of synthetic chemistry and molecular biology techniques.
ArM Construction Methodologies
Four principal strategies have been established for assembling functional ArMs, each offering distinct advantages for specific applications:
Table 1: Primary Strategies for Artificial Metalloenzyme Assembly
| Assembly Strategy | Mechanism | Key Features | Common Applications |
|---|---|---|---|
| Covalent Anchoring | Irreversible chemical bonding between metal complex and protein side chains | Stable conjugation; precise positioning | Cysteine-based linkages; post-translational modifications [11] |
| Supramolecular Anchoring | High-affinity non-covalent interactions | Modular assembly; facile screening | Biotin-streptavidin systems; antibody-antigen recognition [11] [12] |
| Metal Substitution | Replacement of native metal in natural metalloenzyme | Altered electronic/steric properties | Novel catalytic pathways in repurposed natural enzymes [11] [13] |
| Dative Anchoring | Direct coordination of metal by protein amino acid residues | Simple implementation; minimal synthetic modification | Natural amino acid coordination (His, Cys, Glu, Asp) [11] |
A fifth emerging strategy involves the genetic incorporation of metal-chelating unnatural amino acids, which enables precise positioning of metal coordination sites directly within the protein backbone through amber stop codon suppression techniques [11]. This approach provides atomic-level control over the first coordination sphere while maintaining the evolvability of the protein scaffold.
The New-to-Nature Reaction Paradigm
Conceptual Framework
New-to-nature reactions represent chemical transformations that expand beyond the known repertoire of biological catalysis. These reactions leverage reaction mechanisms and catalytic approaches that have not evolved in natural biological systems, effectively creating synthetic metabolic pathways and enabling the production of non-biological compounds [14]. The pursuit of these reactions represents a fundamental shift in biocatalysis, from exploiting nature's existing toolkit to creating entirely new biocatalytic functions.
This paradigm has been particularly powerful in addressing synthetic challenges in pharmaceutical and fine chemical synthesis. For example, the development of an artificial Suzukiase based on biotin-streptavidin technology enables enantioselective Suzuki coupling reactions, a transformation previously restricted to synthetic catalysts [11]. Similarly, ArMs have been engineered to perform olefin metathesis, C-H activation, and cyclopropanation reactions with biological compatibility [11].
Representative Reaction Classes
The reaction scope facilitated by ArMs has expanded dramatically in recent years, encompassing numerous valuable transformations:
Table 2: Categories of New-to-Nature Reactions Catalyzed by Artificial Metalloenzymes
| Reaction Category | Specific Examples | Significance |
|---|---|---|
| Cross-Coupling Reactions | Suzuki reaction [11], Heck reaction [11] | C-C bond formation for pharmaceutical synthesis |
| Carbene/Nitrene Transfer | Cyclopropanation [11], C-H amination [14] | Introduction of stereocenters and strained rings |
| Radical Chemistry | Atom transfer radical cyclization [11], Giese-type radical conjugate addition [15] | Access to challenging radical intermediates under mild conditions |
| Oxidation & Reduction | Asymmetric hydrogenation [11], alcohol oxidation [11] | Selective redox transformations without heavy metal contaminants |
| Multi-Step Cascades | Chemoenzymatic cascades [12] | Streamlined synthesis without intermediate isolation |
The mechanism behind many new-to-nature reactions often involves the generation of highly reactive intermediates, such as metal-carbene or metal-nitrene species, which are subsequently harnessed within the protein scaffold to achieve stereoselective transformations [14]. For instance, engineered cytochrome P450 enzymes can be repurposed to perform abiological carbene transfer reactions that proceed through reactive iron-carbene intermediates, enabling cyclopropanation and other valuable transformations [14].
Experimental Design and Optimization Workflows
Integrated Development Pipeline
The creation of functional ArMs follows an iterative development process that integrates design, assembly, and optimization phases. The workflow typically begins with scaffold selection, where researchers choose appropriate protein frameworks based on structural properties, engineering feasibility, and compatibility with the target reaction. Common scaffolds include streptavidin, multidrug resistance regulators (LmrR), and various β-barrel proteins [11].
Diagram 1: ArM Development Workflow
Following initial assembly, ArMs undergo systematic optimization through directed evolution, a powerful protein engineering approach that mimics natural evolution in laboratory settings. This process involves iterative cycles of mutagenesis and high-throughput screening to identify variants with enhanced activity, selectivity, or stability [14]. The 2018 Nobel Prize in Chemistry awarded to Frances H. Arnold recognized the transformative potential of directed evolution for enzyme engineering, including the optimization of ArMs [11].
Key Methodological Approaches
Directed Evolution Protocol
Library Generation: Create genetic diversity through error-prone PCR or DNA shuffling of the gene encoding the protein scaffold [14].
Expression and Assembly: Express variant proteins in suitable host systems (typically E. coli) and reconstitute with the artificial metal cofactor [11].
High-Throughput Screening: Implement rapid assays to evaluate catalytic performance (activity, enantioselectivity) across thousands of variants [14].
Variant Selection: Identify improved clones and use them as templates for subsequent evolution rounds [14].
This methodology enabled the transformation of cytochrome P450 enzymes with trace C-H amination activity into efficient catalysts capable of hundreds to thousands of turnovers with high stereoselectivity [14].
Photobiocatalysis Development
Recent advances have integrated photoredox catalysis with ArM technology:
Cofactor Excitation: Utilize visible light to excite engineered ketoreductase enzymes, enabling radical generation from alkyl halide precursors [15].
Stereocontrol: Leverage the enzyme active site to control radical intermediate stereochemistry, enabling asymmetric transformations [15].
Reaction Optimization: Fine-tune reaction conditions (wavelength, temperature, substrate loading) to maximize yield and selectivity [15].
This approach has enabled challenging reactions such as asymmetric hydrogen atom transfer and hydroalkylation of styrenes, expanding the synthetic utility of flavin-dependent enzymes beyond their natural two-electron redox chemistry [14].
Research Reagents and Toolkits
Essential Research Materials
The development and application of ArMs relies on specialized reagents and tools that facilitate their construction, optimization, and implementation:
Table 3: Essential Research Reagents for Artificial Metalloenzyme Development
| Reagent Category | Specific Examples | Research Function |
|---|---|---|
| Protein Scaffolds | Streptavidin variants [11], LmrR [11], Cytochrome P450 [14] | Provides evolvable chiral environment for metal cofactor |
| Metal Cofactors | Biotinylated piano-stool complexes [12], Fe(heme) complexes [11], Cu(phenanthroline) complexes [11] | Imparts novel catalytic activity and reaction mechanisms |
| Genetic Tools | Amber stop codon suppression systems [11], Metallo-CRISPR libraries [16] | Enables incorporation of unnatural amino acids and targeted mutagenesis |
| Analytical Methods | Computational docking tools [16], High-throughput screening assays [17] | Facilitates design and optimization through rapid performance evaluation |
| Specialized Libraries | Metal-binding pharmacophores (MBPs) [16], Fragment libraries [18] | Provides building blocks for inhibitor design and cofactor development |
The increasing integration of machine learning and computational design has dramatically accelerated ArM development:
RFdiffusion, a recently developed protein design tool, enables de novo generation of protein backbones tailored to accommodate specific functional motifs [19]. By fine-tuning the RoseTTAFold structure prediction network on protein structure denoising tasks, researchers can generate novel protein scaffolds optimized for metal cofactor incorporation and catalytic function [19].
Complementary tools like CATNIP (Compatibility Assessment Tool for Non-natural Intermediate Partnerships) help predict productive enzyme-substrate pairs for specific transformations, particularly for α-ketoglutarate/Fe(II)-dependent enzyme systems [17]. This predictive capability reduces the experimental burden associated with identifying starting points for enzyme engineering.
Emerging Applications and Future Directions
Therapeutic Development
ArM technology has significant implications for pharmaceutical research and development. Blacksmith Medicines has leveraged metalloenzyme-targeting platforms to develop FG-2101, a novel non-hydroxamate antibiotic that inhibits LpxC—a zinc-dependent metalloenzyme found exclusively in Gram-negative bacteria [16] [18]. This approach addresses the historical challenges associated with targeting metalloenzymes, which represent over 30% of all known enzymes but have proven difficult to drug with conventional small molecules [18].
Sustainable Chemistry and Biomanufacturing
The application of ArMs in industrial biocatalysis offers opportunities for more sustainable manufacturing processes. By enabling efficient chemical synthesis in water under mild conditions, ArMs can reduce energy consumption and waste generation associated with traditional chemical catalysis [15]. Their compatibility with biological systems also facilitates the development of chemoenzymatic cascades, where artificial and natural enzymes work in concert to convert renewable feedstocks into valuable chemicals [12].
Recent advances in intracellular ArM catalysis have demonstrated the potential for implementing new-to-nature reactions within living cells, opening possibilities for synthetic biology and metabolic engineering applications [11] [12]. This capability could enable the microbial production of complex molecules through artificial biosynthetic pathways that incorporate non-biological reaction steps.
Knowledge Gaps and Research Challenges
Despite significant progress, several challenges remain in the field of artificial metalloenzymes. The predictable integration of non-biological cofactors into protein scaffolds continues to require substantial optimization, and the general rules governing second-sphere interactions in ArMs are not fully understood [13]. Additionally, the scalability of ArM-catalyzed processes for industrial applications needs further demonstration, particularly for complex multi-step transformations.
Future research directions will likely focus on expanding the reaction scope of ArMs, improving computational design accuracy, and developing more efficient strategies for optimizing ArM performance. The integration of machine learning approaches with high-throughput experimental validation represents a particularly promising avenue for accelerating the development cycle [14] [19]. As these tools mature, artificial metalloenzymes are poised to become increasingly powerful catalysts for solving challenging problems in synthetic chemistry and biotechnology.
The de novo design of novel enzyme functions represents a frontier in synthetic biology, aiming to create tailored biocatalysts that operate with high efficiency in demanding industrial and therapeutic environments. The success of these designed enzymes hinges on achieving three critical design objectives: thermostability, solvent tolerance, and cofactor compatibility. Thermostability ensures enzymatic integrity and function at elevated temperatures, accelerating reaction rates and preventing aggregation. Solvent tolerance enables functionality in non-aqueous environments essential for industrial biocatalysis where substrate solubility is limited. Cofactor compatibility expands catalytic repertoire by incorporating synthetic metal complexes and abiotic cofactors to catalyze "new-to-nature" reactions. This technical guide examines the fundamental principles, experimental methodologies, and computational frameworks for achieving these design objectives, providing researchers with actionable strategies for advancing de novo enzyme design.
Thermostability: Engineering Robust Molecular Scaffolds
Fundamental Principles and Molecular Strategies
Thermostability is crucial for industrial enzyme applications, directly influencing catalytic efficiency, half-life, and operational costs. Enhancing an enzyme's ability to maintain its native conformation under elevated temperatures involves strategic reinforcement of its structural framework through multiple molecular mechanisms [20].
Short-loop engineering has emerged as a powerful strategy for enhancing thermal stability by targeting rigid "sensitive residues" in short-loop regions. This approach involves mutating these residues to hydrophobic amino acids with large side chains to fill internal cavities, thereby enhancing structural integrity [20]. Unlike traditional B-factor strategies that target flexible regions, short-loop engineering focuses on stabilizing inherently rigid areas that may contain destabilizing cavities. The strategy proved effective across multiple enzyme classes, increasing the half-life of lactate dehydrogenase from Pediococcus pentosaceus by 9.5-fold, urate oxidase from Aspergillus flavus by 3.11-fold, and D-lactate dehydrogenase from Klebsiella pneumoniae by 1.43-fold compared to wild-type enzymes [20].
Hydrophobic core packing represents another crucial mechanism, where clustering hydrophobic residues in the protein core minimizes structural voids and enhances stability. Thermophilic proteins naturally employ this strategy, exhibiting a higher proportion of hydrophobic and charged residues that create a densely packed interior [20]. Computational analyses reveal that cavity-filling mutations can reduce void volumes from 265 ų to less than 48 ų, significantly improving structural rigidity without introducing new hydrogen bonds or salt bridges [20].
Secondary stabilization through hydrogen bonding, salt bridges, and disulfide bonds provides additional stabilization. While not the primary focus of cavity-filling strategies, these elements contribute significantly to overall structural integrity, particularly when strategically positioned to restrict structural "wobble" at high temperatures [20].
Experimental Protocols and Assessment Methods
Virtual Saturation Mutagenesis with Free Energy Calculations: This protocol identifies stabilization sites through computational screening:
- Step 1: Identify short-loop regions (typically 3-8 residues) connecting secondary structural elements
- Step 2: Perform virtual saturation mutagenesis using tools like FoldX to calculate unfolding free energy changes (ΔΔG)
- Step 3: Identify "sensitive residues" where mutations yield negative ΔΔG values, indicating stabilization potential
- Step 4: Prioritize residues with small side chains (e.g., alanine) creating cavities for mutation to larger hydrophobic residues (phenylalanine, tyrosine, tryptophan, methionine)
Experimental Validation Pipeline:
- Library Construction: Generate saturation mutagenesis libraries for identified sensitive residues
- Expression Screening: Express variants and assess solubility and folding
- Thermal Stability Assays:
- Determine half-life (t₁/₂) at elevated temperatures
- Measure melting temperature (Tₘ) using differential scanning fluorimetry
- Calculate residual activity after incubation at target temperatures
- Structural Analysis:
- Perform molecular dynamics simulations to assess root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF)
- Analyze cavity volumes pre- and post-mutation
- Identify formation of new stabilizing interactions
Table 1: Quantitative Improvements in Enzyme Thermostability via Short-Loop Engineering
| Enzyme | Source Organism | Mutation | Half-life Improvement (Fold) | Key Mechanism |
|---|---|---|---|---|
| Lactate Dehydrogenase | Pediococcus pentosaceus | A99Y | 9.5 | Cavity filling, enhanced hydrophobic interactions |
| Urate Oxidase | Aspergillus flavus | Not specified | 3.11 | Cavity filling, structural compaction |
| D-Lactate Dehydrogenase | Klebsiella pneumoniae | Not specified | 1.43 | Cavity filling, hydrophobic clustering |
Solvent Tolerance: Designing for Non-Aqueous Environments
Molecular Adaptations for Organic Solvents
Industrial biocatalysis often requires operation in non-aqueous environments where organic solvents are necessary for substrate solubility or product recovery. Solvent tolerance encompasses an enzyme's ability to maintain structural integrity and catalytic activity in the presence of organic solvents, which typically strip essential water molecules, disrupt hydrogen bonds, and cause structural denaturation.
Surface charge engineering enhances solvent tolerance by optimizing surface charge distribution to maintain hydration layers in organic solvents. Introducing charged residues (glutamate, aspartate, lysine, arginine) on the protein surface strengthens protein-solvent interactions and prevents aggregation in low-dielectric environments [21]. Rational design of surface charges can be guided by computational tools that model protein-solvent interactions and identify regions prone to destabilization.
Surface hydrophobization represents a counterintuitive yet effective strategy where increasing surface hydrophobicity improves compatibility with organic solvents. This approach reduces the energetic penalty of transferring the enzyme from aqueous to organic environments and prevents unfavorable interactions at the protein-solvent interface [21]. Strategic mutation of polar surface residues to hydrophobic ones (leucine, valine, isoleucine) can significantly enhance stability in organic media.
Structural rigidification through the introduction of disulfide bonds and proline residues reduces conformational flexibility, minimizing unfolding in dehydrating environments. Computational tools like RosettaDesign can identify potential disulfide bond formation sites that stabilize the native state without compromising catalytic function [19].
Experimental Assessment of Solvent Tolerance
Solvent Stability Assays:
- Incubation Protocol: Incubate enzymes in water-solvent mixtures (e.g., 25% DMSO, 30% methanol, 20% acetonitrile) for predetermined durations
- Activity Measurements: Assess residual activity using standard enzymatic assays compared to aqueous controls
- Structural Integrity:
- Circular dichroism spectroscopy to monitor secondary structural changes
- Fluorescence spectroscopy to analyze tertiary structural alterations
- Dynamic light scattering to detect aggregation
Solvent Tolerance Screening Pipeline:
- Primary Screening: High-throughput assessment of activity in microtiter plates with various solvent conditions
- Secondary Validation: Detailed kinetic analysis (Kₘ, kcat) in optimal solvent systems
- Tertiary Characterization: Long-term stability studies under operational conditions
Table 2: Strategic Approaches for Enhancing Enzyme Solvent Tolerance
| Strategy | Molecular Approach | Experimental Implementation | Expected Outcome |
|---|---|---|---|
| Surface Charge Engineering | Introduce charged residues at solvent-exposed positions | Computational surface analysis followed site-directed mutagenesis | Improved hydration layer maintenance in polar solvents |
| Surface Hydrophobization | Replace polar surface residues with hydrophobic counterparts | Saturation mutagenesis of surface residues followed by solvent screening | Enhanced stability in non-polar organic solvents |
| Structural Rigidification | Introduce disulfide bonds or proline residues at flexible loops | Computational design of stabilizing disulfides with geometric constraints | Reduced conformational flexibility and unfolding in dehydrating environments |
Cofactor Compatibility: Expanding Catalytic Repertoire
Designing Artificial Metalloenzymes
Cofactor compatibility addresses the challenge of incorporating synthetic metal complexes and abiotic cofactors into protein scaffolds to catalyze non-biological reactions. This objective represents the cutting edge of de novo enzyme design, enabling chemical transformations beyond nature's repertoire [5].
The de novo design of artificial metalloenzymes (ArMs) requires creating tailored protein scaffolds that can bind synthetic cofactors while providing an optimal environment for catalysis. Recent breakthroughs include the development of an artificial metathase for ring-closing metathesis reactions in cellular environments [5]. This approach combines computational design with genetic optimization to achieve high binding affinity (K_D ≤ 0.2 μM) between the protein scaffold and cofactor through supramolecular anchoring [5].
Supramolecular anchoring strategies enable precise positioning of metal cofactors within designed protein pockets. Unlike covalent attachment, supramolecular interactions allow for cofactor exchange and tuning of the catalytic environment. The design process involves:
- Identifying complementary interaction surfaces between cofactor and protein
- Engineering hydrophobic pockets for cofactor binding
- Incorporating hydrogen bond donors/acceptors for precise orientation
- Creating steric constraints to shield reactive intermediates
Scaffold selection criteria for ArM design prioritize hyperstable de novo-designed proteins with engineered binding sites rather than repurposing natural scaffolds. These designs offer enhanced tunability and stability, enabling function in complex cellular environments [5]. The closed alpha-helical toroidal repeat proteins (dnTRPs) have proven particularly effective due to their high thermostability (T₅₀ > 98°C) and engineerability [5].
Protocol for Artificial Metathase Design and Optimization
Computational Design Pipeline:
- Step 1: Cofactor Design: Modify synthetic cofactors to include polar motifs for specific interactions with protein scaffolds (e.g., sulfamide groups for hydrogen bonding)
- Step 2: Scaffold Design: Use computational suites (RifGen/RifDock) to enumerate interacting amino acid rotamers and dock ligands into de novo protein cavities
- Step 3: Sequence Optimization: Employ Rosetta FastDesign to optimize hydrophobic contacts and stabilize key hydrogen-bonding residues
- Step 4: Binding Affinity Enhancement: Introduce tryptophan residues near the binding site to enhance hydrophobicity and cofactor affinity
Directed Evolution Protocol:
- Library Generation: Create mutant libraries targeting residues surrounding the cofactor binding pocket
- Screening Conditions: Develop cell-free extract screening systems supplemented with additives (e.g., Cu(Gly)₂) to mitigate glutathione interference
- Performance Assessment: Evaluate variants based on turnover number (TON) and biocompatibility
- Iterative Optimization: Perform multiple rounds of mutation and screening to achieve significant catalytic improvements (≥12-fold enhancement documented) [5]
Table 3: Performance Metrics for Artificial Metathase Design
| Design Stage | Key Parameter | Initial Performance | Optimized Performance | Assessment Method |
|---|---|---|---|---|
| Cofactor Binding | Dissociation Constant (K_D) | 1.95 ± 0.31 μM | ≤0.2 μM | Tryptophan fluorescence quenching |
| Catalytic Efficiency | Turnover Number (TON) | 40 ± 4 | ≥1,000 | Product formation rate in cell-free extracts |
| Thermal Stability | T₅₀ (30 min incubation) | Not applicable | >98°C | Temperature-dependent unfolding |
Integrated Computational-Experimental Workflows
AI-Driven De Novo Enzyme Design
Artificial intelligence has revolutionized de novo enzyme design by enabling precise, from-scratch prediction of enzyme structures with tailored functions [4]. Generative AI models have demonstrated remarkable success in creating entirely novel enzyme folds distinct from natural proteins, exemplified by the design of a de novo serine hydrolase with catalytic efficiencies (kcat/Km) up to 2.2 × 10⁵ M⁻¹·s⁻¹ [22].
RFdiffusion represents a groundbreaking approach that fine-tunes the RoseTTAFold structure prediction network for protein structure denoising tasks [19]. This generative model enables unconditional and topology-constrained protein monomer design, protein binder design, symmetric oligomer design, and enzyme active site scaffolding. The method experimentally demonstrated the capacity to design diverse functional proteins from simple molecular specifications, with characterization of hundreds of designed symmetric assemblies, metal-binding proteins, and protein binders confirming design accuracy [19].
Theozyme-Based Design implements an "inside-out" strategy where catalytic sites are designed first by modeling the transition state of the target reaction [22]. Quantum mechanical calculations identify optimal arrangements of catalytic groups to stabilize transition states, creating theoretical enzyme models ("theozymes") that serve as blueprints for subsequent scaffold design. This approach has matured through tools like RosettaMatch, which places theozyme-derived catalytic motifs into protein backbones [22].
Consensus Structure Identification
Complementing theozyme approaches, consensus structure identification employs data-driven strategies to extract conserved geometrical features from natural enzyme families [22]. Analyzing structural databases like the Protein Data Bank reveals conserved spatial relationships and hydrogen-bonding networks associated with catalytic function. This method successfully identifies key catalytic motifs like the serine hydrolase triad (Ser-His-Asp) and associated oxyanion holes, providing evolutionary-validated templates for enzyme design [22].
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 4: Essential Research Tools for Advanced Enzyme Design
| Tool Category | Specific Tools/Platforms | Primary Function | Application Examples |
|---|---|---|---|
| Structure Prediction & Validation | AlphaFold2, RoseTTAFold, ESMFold | Protein structure prediction from sequence | Validation of de novo enzyme designs, structural analysis |
| Generative Design Platforms | RFdiffusion, GENzyme, SCUBA-D | De novo protein backbone generation | Creating novel enzyme scaffolds around functional motifs |
| Sequence Design Tools | ProteinMPNN, LigandMPNN | Inverse protein folding for sequence design | Optimizing sequences for target structures and cofactor binding |
| Molecular Modeling & Simulation | Rosetta, FoldX, GROMACS | Energy calculations, docking, dynamics | Virtual mutagenesis, stability predictions, binding affinity |
| Quantum Chemistry Software | Gaussian, ORCA, Q-Chem | Transition state modeling, theozyme design | Catalytic mechanism analysis, active site optimization |
| Directed Evolution Systems | Cell-free expression, microfluidics | High-throughput screening of enzyme variants | Optimization of initially designed enzymes for enhanced function |
| Biophysical Characterization | SPR, ITC, CD, fluorescence spectroscopy | Binding affinity, structural stability | Validation of cofactor binding, thermal stability assessment |
The integration of advanced computational design with experimental optimization has transformed enzyme engineering from an art to a predictive science. The key objectives of thermostability, solvent tolerance, and cofactor compatibility represent interconnected challenges that must be addressed simultaneously for successful de novo enzyme design. Short-loop engineering and cavity-filling strategies provide robust approaches for enhancing thermostability, while surface engineering techniques enable operation in non-aqueous environments. Most remarkably, the de novo creation of artificial metalloenzymes demonstrates the potential to expand catalytic repertoire beyond natural evolution, enabling abiotic chemistry in biological systems.
Future advances will likely emerge from increasingly sophisticated AI models trained on expanding structural databases, improved quantum mechanical methods for modeling reaction mechanisms, and high-throughput experimental characterization that provides feedback for computational refinement. As these technologies mature, the precise design of enzymes with tailored stability, solvent compatibility, and catalytic functions will accelerate progress in sustainable chemistry, therapeutic development, and synthetic biology.
The field of de novo enzyme design aims to create novel biocatalysts from first principles, expanding the repertoire of biological catalysis to include non-natural reactions. A fundamental challenge in this endeavor is the successful incorporation of artificial metal cofactors—the abiotic catalytic centers that enable new-to-nature functions. The strategy used to anchor these cofactors within protein scaffolds directly determines the stability, activity, and biocompatibility of the resulting artificial metalloenzyme (ArM). Researchers primarily employ three strategic approaches: supramolecular anchoring (utilizing non-covalent interactions), covalent anchoring (forming chemical bonds), and dative anchoring (leveraging metal-coordination bonds) [23]. Within the context of de novo design, where protein scaffolds are computationally conceived rather than naturally evolved, the choice of anchoring strategy profoundly influences the design process, the final catalytic efficiency, and the potential for in-cellulo applications. This technical guide examines these core anchoring strategies, their implementation, and their integration into the broader framework of designing novel enzyme functions.
Supramolecular Anchoring
Principle and Strategic Value
Supramolecular anchoring relies on non-covalent interactions—such as hydrogen bonding, hydrophobic effects, and π-π interactions—to embed a synthetic cofactor within a protein binding pocket [23]. This approach is particularly valuable in de novo design, as it allows designers to treat the cofactor and the protein as two separate modules. The design process can thus focus on creating a pocket with complementary geometry and chemical properties to the cofactor, without the constraints of designing specific covalent attachment points. A key advantage is the potential for cofactor exchange or replacement, facilitating screening and optimization. However, a potential drawback is the risk of cofactor leaching, especially under dilute conditions or in dynamic cellular environments.
Implementation in De Novo Design
A prominent example of this strategy is the creation of an artificial metathase for ring-closing metathesis. Researchers designed a de novo hyper-stable alpha-helical toroidal repeat protein (dnTRP) scaffold to host a tailored Hoveyda-Grubbs ruthenium catalyst (Ru1) [23]. The design process involved computational docking of the Ru1 cofactor into the scaffold's cavity, explicitly designing the binding pocket to provide supramolecular anchoring via:
- Hydrogen bonds between the protein backbone and the polar sulfamide group of the Ru1 cofactor.
- Hydrophobic interactions with the cofactor's mesityl moieties.
This designed supramolecular interface achieved a high binding affinity (KD ≤ 0.2 μM), demonstrating that de novo proteins can be engineered to tightly bind abiotic cofactors without covalent or dative links [23].
Table 1: Key Characteristics of Cofactor Anchoring Strategies
| Anchoring Strategy | Interaction Type | Design Complexity | Binding Strength | Risk of Cofactor Leaching | Ease of Cofactor Incorporation |
|---|---|---|---|---|---|
| Supramolecular | Non-covalent (H-bond, hydrophobic) | High (requires precise pocket design) | Moderate to Strong (nM-μM KD) | Moderate | High |
| Covalent | Covalent bond | Moderate (requires addressable residues) | Strong (Irreversible) | Low | Low to Moderate |
| Dative | Metal coordination | Moderate (requires coordinating residues) | Strong | Low | Moderate |
Experimental Protocol: Measuring Binding Affinity via Tryptophan Fluorescence Quenching
Objective: To determine the dissociation constant (KD) for a supramolecularly bound cofactor-protein complex.
- Protein Preparation: Express and purify the de novo designed protein (e.g., dnTRP_18) with a fluorescent residue (e.g., Tryptophan) positioned near the binding pocket.
- Sample Preparation: Prepare a series of protein solutions at a fixed concentration (e.g., 1 μM) in a suitable buffer (e.g., pH 4.2).
- Titration: Titrate increasing concentrations of the cofactor (e.g., Ru1) into the protein solution.
- Fluorescence Measurement: After each addition, measure the fluorescence emission intensity of the tryptophan upon excitation at 295 nm.
- Data Analysis: Plot the measured fluorescence intensity (or quenching efficiency) against the cofactor concentration. Fit the data to a binding isotherm model (e.g., one-site specific binding) to calculate the KD value [23].
Covalent and Dative Anchoring Strategies
Covalent Anchoring
Covalent anchoring involves the formation of irreversible chemical bonds between the protein scaffold and the synthetic cofactor. This is often achieved by reacting engineered cysteine residues (thiol groups) with functional groups like maleimides or iodoacetamides on the cofactor [23]. The primary advantage of this method is the exceptional complex stability it confers, virtually eliminating cofactor leaching and making it suitable for harsh reaction conditions. A significant disadvantage is that the bond formation can be challenging to perform in living cells, and the fixed attachment point may restrict conformational dynamics necessary for optimal catalysis.
Dative Anchoring
Dative anchoring, or metal coordination, utilizes the native ligating atoms of protein side chains (e.g., His, Cys, Asp, Glu) to coordinate directly to a metal center in the cofactor [23]. This strategy mimics the cofactor binding in many natural metalloenzymes. It provides strong, directional binding, though the bond is potentially reversible. The design process involves positioning coordinating residues in the scaffold's active site to match the geometric constraints of the metal cofactor. While this can yield very active ArMs, a major challenge is the potential for mis-metalation in a cellular environment, where endogenous metal ions can compete for the binding site.
Table 2: Comparison of Anchoring Strategy Performance in Artificial Metalloenzymes
| Performance Metric | Supramolecular | Covalent | Dative |
|---|---|---|---|
| Reported Turnover Number (TON) | ≥ 1,000 [23] | Varies (often high) | Varies (often high) |
| Stability in Complex Media | High (with optimized binding) | Very High | High (subject to metal competition) |
| In Cellulo Compatibility | Demonstrated [23] | Can be challenging | Can be challenging |
| Directed Evolution Friendliness | High (scaffold can be evolved independently) | Moderate | Moderate |
Integration with De Novo Enzyme Design Workflows
The creation of a functional ArM is an iterative process that integrates anchoring strategy with computational design and experimental optimization. The following workflow diagram illustrates the generic pathway for developing an ArM, which can be tailored for any of the three anchoring strategies.
ArM Development Workflow
Computational Design and Experimental Validation
The initial phase involves computational design of the protein scaffold. For supramolecular anchoring, tools like Rosetta and the RifGen/RifDock suite are used to enumerate amino acid rotamers around the cofactor and dock it into de novo scaffolds (e.g., dnTRPs) [23]. The design is evaluated on metrics like interface quality and pocket pre-organization. The selected designs are then expressed, purified, and assembled with the cofactor.
Catalytic performance is tested under relevant conditions. For example, artificial metathases were tested for ring-closing metathesis activity with a diallylsulfonamide substrate [23]. Key analytical methods include:
- Chromatography (e.g., LC-MS/GC): To quantify substrate conversion and product formation.
- Native Mass Spectrometry: To confirm 1:1 cofactor:protein stoichiometry.
- Size-Exclusion Chromatography: To verify complex formation and stability.
Optimization via Directed Evolution
Even with sophisticated computational design, initial ArMs often require optimization. Directed evolution is a powerful method for this, where iterative cycles of mutagenesis and high-throughput screening are used to enhance catalytic performance (e.g., TON, enantioselectivity) and biocompatibility [23] [24]. This process can improve the activity of a designed ArM by more than 12-fold, making it compatible with complex environments like bacterial cytoplasm [23]. Screening can be performed in cell-free extracts (CFE) under optimized conditions, such as adjusted pH and the addition of additives like bis(glycinato)copper(II) to mitigate the effects of cellular metabolites like glutathione [23].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Developing Artificial Metalloenzymes
| Reagent / Material | Function / Application | Example Use Case |
|---|---|---|
| De Novo Protein Scaffolds | Provides a stable, customizable framework for cofactor binding. | Hyper-stable dnTRP scaffolds for supramolecular anchoring [23]. |
| Hoveyda-Grubbs Catalyst Derivatives | Abiotic cofactor for olefin metathesis reactions. | Ru1 catalyst for artificial metathase design [23]. |
| E. coli Expression Systems | Standard host for recombinant protein production. | Expression of his-tagged dnTRP proteins [23]. |
| Rosetta Software Suite | Computational protein design and modeling. | Designing and optimizing cofactor-binding pockets [23] [3]. |
| Cell-Free Extracts (CFE) | Mimics the intracellular environment for screening. | High-throughput screening of ArM variants in a biologically complex medium [23]. |
| Bis(glycinato)copper(II) [Cu(Gly)₂] | Additive to mitigate reducing environments in lysates. | Oxidation of glutathione in CFE to protect ruthenium cofactors [23]. |
The strategic selection of an anchoring method—supramolecular, covalent, or dative—is a foundational decision in the de novo design of artificial metalloenzymes. Supramolecular strategies offer a modular and design-friendly approach that has proven highly successful for creating ArMs functional in living cells. Covalent and dative strategies provide robust stability, though they present different challenges for in-cellulo implementation. The integration of sophisticated computational design, leveraging tools like Rosetta and machine learning, with powerful experimental optimization techniques like directed evolution, creates a robust framework for advancing the field. As computational methods continue to improve, the precision with which cofactor environments can be designed will increase, further enabling the creation of efficient and selective biocatalysts for a wide range of abiotic transformations in both industrial and biomedical contexts.
Artificial metalloenzymes (ArMs) present a promising avenue for abiotic catalysis within living systems. However, their in vivo application is currently limited by critical challenges, particularly in selecting suitable protein scaffolds capable of binding abiotic cofactors and maintaining catalytic activity in complex media. This case study details a pronounced leap in the de novo design and in cellulo engineering of an artificial metathase—an ArM designed for ring-closing metathesis (RCM) for whole-cell biocatalysis. The approach integrates a tailored metal cofactor into a hyper-stable, de novo-designed protein. By combining computational design with genetic optimization, a high binding affinity (KD ≤ 0.2 μM) between the protein scaffold and cofactor was achieved through supramolecular anchoring. Directed evolution of the artificial metathase yielded variants exhibiting excellent catalytic performance (turnover number ≥1,000) and biocompatibility, paving the way for abiological catalysis in living systems [5] [25].
The field of biocatalysis is increasingly attractive for synthetic chemistry due to its benefits in sustainability, step economy, and exquisite selectivity. A frontier in this field is the creation of artificial metalloenzymes (ArMs), which aim to merge the catalytic versatility of synthetic metal complexes with the advantageous performance of enzymes in biological environments [5]. A primary goal is to catalyze "new-to-nature" reactions—transformations with no equivalent in natural biology—within living cells [26].
Olefin metathesis, a reaction for which the 2005 Nobel Prize in Chemistry was awarded, is one such powerful transformation. It enables the rearrangement of carbon-carbon double bonds and is widely used in organic synthesis and materials science [27]. Despite its utility, the application of olefin metathesis in chemical biology has been limited because conventional ruthenium catalysts often suffer from poor biocompatibility, instability in aqueous media, and deactivation by cellular metabolites like glutathione [5].
This case study, framed within a broader thesis on the de novo design of novel enzyme functions, examines a groundbreaking solution to these challenges. It chronicles the rational design and evolution of an artificial metathase, demonstrating the feasibility of performing abiotic catalysis in the complex cytoplasmic environment of E. coli.
Computational De Novo Design of the Host Protein
Design Strategy and Rationale
The design strategy hinged on a synergistic approach: engineering both a synthetic cofactor and a de novo-designed protein scaffold to complement each other [5].
- Cofactor Design: A derivative of the Hoveyda-Grubbs catalyst, termed Ru1, was synthesized. A key feature was the incorporation of a polar sulfamide group, intended to improve aqueous solubility and serve as a handle for forming supramolecular interactions (e.g., hydrogen bonds) with the host protein [5].
- Scaffold Selection: De novo-designed closed alpha-helical toroidal repeat proteins (dnTRPs) were selected as the scaffold. These proteins are hyper-stable (T50 > 98°C), highly engineerable, and possess a suitably sized pocket for ligand binding, making them ideal for withstanding the rigors of cytoplasmic catalysis and engineering [5].
Computational Methodology and Workflow
The process for designing the host protein involved a multi-stage computational pipeline [5] [28]:
- Rotamer Interaction Field (RIF) Generation: The
RifGentool was used to enumerate potential interacting amino acid rotamers around the cofactor, Ru1. - Ligand Docking: The
RifDocksuite was employed to dock the Ru1 cofactor along with key interacting residues into the cavities of pre-existing dnTRP scaffolds (e.g., PDB ID: 4YXX). - Sequence Optimization: The docked structures were subjected to protein sequence optimization using Rosetta FastDesign. This step refined hydrophobic contacts with the cofactor and stabilized key H-bonding residues to pre-organize the binding pocket for catalysis.
- Design Selection: The resulting design models were evaluated based on computational metrics describing the protein-cofactor interface and pocket pre-organization. This process yielded 21 initial designs for experimental validation [5].
The following diagram illustrates this integrated computational design workflow:
Experimental Validation and Optimization
Initial Screening and Binding Affinity Improvement
The 17 soluble dnTRPs were purified and assembled into ArMs by treatment with the Ru1 cofactor. Their catalytic performance was assessed using the RCM of diallylsulfonamide (1a) as a model reaction [5].
- Primary Screen: All Ru1·dnTRP complexes outperformed the free Ru1 cofactor (TON 40 ± 4). The best performers, dnTRP10, dnTRP17, and dnTRP_18, achieved TONs of approximately 180-194 [5].
- Lead Selection: dnTRP_18 was selected for further study due to its high activity and robust expression [5].
- Affinity Engineering: To improve the binding affinity, two residues (F43 and F116) lining the binding pocket were individually mutated to tryptophan. The resulting variants, dnTRP18F43W and dnTRP18F116W, showed a nearly tenfold increase in affinity, with KD values of 0.26 ± 0.05 μM and 0.16 ± 0.04 μM, respectively. The dnTRP18F116W variant was designated dnTRP_R0 for subsequent evolution campaigns [5].
Table 1: Key Characterization Data for Lead Artificial Metathase Designs
| Protein Variant | Binding Affinity (KD, μM) | Catalytic Performance (TON) | Key Characteristics |
|---|---|---|---|
| Free Ru1 Cofactor | Not Applicable | 40 ± 4 | Baseline activity in buffer |
| Ru1·dnTRP_18 | 1.95 ± 0.31 | 194 ± 6 | Initial lead design |
| Ru1·dnTRP_R0 (F116W) | 0.16 ± 0.04 | ~200 (Parental) | High-affinity variant, used for directed evolution |
| Evolved Ru1·dnTRP | Not Reported | ≥ 1,000 | Post-directed evolution performance |
Directed Evolution in a Cellular Environment
To optimize the ArM for function in biologically relevant conditions, a directed evolution campaign was initiated. A key development was the establishment of a screening system using E. coli cell-free extracts (CFE) to mimic the cytoplasmic environment [5].
- Screening Platform: The CFE system was supplemented with bis(glycinato)copper(II) [Cu(Gly)2], which partially oxidizes glutathione, a key cellular nucleophile that can deactivate the ruthenium cofactor. This step was critical for achieving high TONs (197 ± 7 with Cu(Gly)2 vs. 152 ± 16 without) in the complex media [5].
- Evolution Strategy: Using this screening platform, iterative rounds of mutagenesis and screening were performed on the dnTRP_R0 scaffold. This process yielded evolved variants with a ≥12-fold increase in catalytic performance compared to the initial designs, achieving TONs of ≥1,000 [5].
The overall experimental workflow, from initial screening to evolved catalyst, is summarized below:
The Scientist's Toolkit: Essential Research Reagents
The development and application of the artificial metathase relied on a suite of key reagents and methodologies.
Table 2: Key Research Reagent Solutions for Artificial Metathase Engineering
| Reagent / Tool | Function and Role in the Study |
|---|---|
| Ru1 Cofactor | A tailored Hoveyda-Grubbs type catalyst with a polar sulfamide group; the abiotic catalytic center of the ArM [5]. |
| dnTRP Scaffold | A hyper-stable, de novo-designed alpha-helical repeat protein; provides a stable, engineerable host for the cofactor [5]. |
| Rosetta Software Suite | A computational protein design platform; used for sequence optimization and binding pocket design around the Ru1 cofactor [5] [28]. |
| RifGen / RifDock | Computational tools for generating rotamer interaction fields and docking small molecules into protein scaffolds [5] [28]. |
| E. coli Cell-Free Extract (CFE) | A complex lysate used for screening; mimics the cytoplasmic environment to identify variants with robust biocompatibility and activity [5]. |
| Bis(glycinato)copper(II) [Cu(Gly)2] | A glutathione-oxidizing agent; added to screening assays to mitigate catalyst deactivation by cellular nucleophiles [5]. |
This case study exemplifies the power of integrating computational design with directed evolution to create novel biocatalysts. The successful development of an artificial metathase for cytoplasmic olefin metathesis underscores several critical advances:
- Synergistic Design: The concurrent engineering of the cofactor and the protein scaffold led to a system with high intrinsic affinity and activity.
- Stability as a Key Enabler: The use of a hyper-stable de novo scaffold provided a robust platform that could withstand the demands of both evolution and the cellular environment.
- Relevant Screening Conditions: The implementation of a screening system in cell-free extracts was pivotal for optimizing the ArM for performance in a complex, biologically relevant milieu.
This work provides a versatile blueprint for creating and optimizing ArMs for a wide range of abiological reactions, significantly expanding the toolbox for synthetic biology and pharmaceutical development. Future work will likely focus on expanding the reaction scope of de novo-designed ArMs and further improving their catalytic efficiency and specificity through advanced computational models and machine learning approaches [29] [30].
The Designer's Toolkit: Integrated Workflows from Computational Prediction to In Vivo Application
The de novo design of novel enzyme functions represents a paradigm shift in biotechnology, moving beyond the modification of existing natural enzymes to the computational creation of entirely new protein scaffolds from first principles. This approach allows researchers to address fundamental scientific questions and engineer biocatalysts for reactions not found in nature, overcoming the limitations of natural enzymes, which often exhibit narrow operating conditions, limited stability, or insufficient activity for industrial applications [3] [31]. Computational scaffolding is the cornerstone of this process, wherein stable protein backbones are designed in silico to precisely position catalytic residues and cofactors for optimal function.
This technical guide examines the core methodologies—Rosetta, RifDock, and emerging deep-learning tools—for constructing de novo protein scaffolds. It details their underlying principles, provides actionable experimental protocols, and situates them within the broader context of functional enzyme design, providing researchers and drug development professionals with the foundational knowledge to implement these cutting-edge strategies.
Core Principles of Computational Scaffolding
Computational scaffolding aims to create a stable, minimal, and designable protein structure that can host a predefined functional motif. Two primary strategies dominate the field:
- Structure-Based Design: This approach uses physical energy functions and spatial pattern algorithms to derive stable protein conformations from three-dimensional constraints. It relies on principles of energetic stabilization and shape complementarity to build scaffolds de novo [3] [32].
- Sequence-Based Design: This strategy employs deep generative models, trained on large datasets of natural protein sequences and structures, to learn co-evolutionary patterns and generate novel, functional sequences from data-driven principles [3].
A key concept in de novo enzyme design is the "inside-out" strategy, which begins by defining the functional site. A minimal active site model, or theozyme (theoretical enzyme), is constructed using quantum mechanical (QM) calculations to identify the optimal spatial arrangement of catalytic residues for stabilizing the reaction's transition state [22]. The computational challenge is then to design a novel protein scaffold that can fold and structurally support this theozyme with atomic-level precision.
Key Methodologies and Tools
The Rosetta Software Suite
Rosetta is a foundational suite of algorithms for de novo protein design and structure prediction. Its methodologies are grounded in physicochemical principles and fragment-based assembly.
- Fundamental Principles: Rosetta uses a Monte Carlo approach to sample conformational space, guided by a physically derived energy function that favors low-energy, stable states. This function balances terms for van der Waals interactions, solvation, hydrogen bonding, and electrostatics [3].
- Key Protocols:
- Motif Placement (RosettaMatch): The functional motif (theozyme) is positioned into a large library of protein backbone scaffolds. The algorithm identifies locations where the catalytic geometry can be accommodated without steric clashes [22].
- Sequence Design (FastDesign): Once a scaffold and motif placement are selected, Rosetta's FastDesign protocol optimizes the amino acid sequence to stabilize both the overall fold and the functional site. This involves iterative cycles of side-chain repacking and backbone minimization [5].
- Application in Scaffolding: Rosetta has been used to design entire protein folds, such as triosephosphate isomerase (TIM) barrels. By revisiting basic topology principles, researchers used Rosetta to create oval-shaped TIM barrels, which are more suitable for incorporating small-molecule binding sites than naturally occurring circular barrels [32].
The RifDock Platform
RifDock is a specialized tool within the Rosetta ecosystem for high-throughput docking of small molecules into protein scaffolds, crucial for designing artificial metalloenzymes (ArMs).
- Fundamental Principles: RifDock leverages the Rotamer Interaction Field (Rif) approach. It pre-computes and enumerates vast libraries of amino acid side-chain rotamers and their favorable interactions with a target small molecule or cofactor. These interaction pairs are then docked into potential binding pockets of protein scaffolds [5].
- Key Protocols:
- Interaction Enumeration: RifDock generates a set of "Rif residues"—rotamers that form specific, favorable interactions (e.g., H-bonds, pi-stacking) with the cofactor.
- Docking into De Novo Scaffolds: These Rif residues and the cofactor are docked as a rigid body into the cavities of stable, de novo-designed protein scaffolds, such as helical bundles.
- Sequence Optimization: The surrounding protein sequence is optimized using Rosetta to improve hydrophobic contacts and stabilize key interacting residues, resulting in a pre-organized binding pocket [5].
- Application in Scaffolding: A landmark study demonstrated RifDock's power by designing a hyper-stable, de novo closed alpha-helical toroidal repeat protein (dnTRP) to bind a synthetic Hoveyda-Grubbs ruthenium cofactor. This created an artificial metathase that performed ring-closing metathesis in the cytoplasm of E. coli [5].
AI-Driven Generative Methods
Recent advances in deep learning have introduced powerful generative models that have revolutionized the scaffolding process.
- RFdiffusion: Fine-tuned from the RoseTTAFold structure prediction network, RFdiffusion is a denoising diffusion probabilistic model that generates protein backbones from noise. It can be conditioned on functional motifs, enabling the de novo generation of protein structures built around a specified active site [19].
- ProteinMPNN: Following backbone generation with tools like RFdiffusion, ProteinMPNN is used for inverse folding. It designs amino acid sequences that are most likely to fold into the generated backbone structure, greatly increasing the experimental success rate of de novo designs [19].
Table 1: Comparison of Core Computational Scaffolding Tools
| Tool | Primary Methodology | Key Function | Strengths | Ideal Use Case |
|---|---|---|---|---|
| Rosetta | Physicochemical energy functions & fragment assembly | Scaffold design, sequence optimization, motif placement | High interpretability, atomic-level control, well-validated | Designing scaffolds for complex functions like TIM barrels |
| RifDock | Rotamer Interaction Field (Rif) & docking | High-throughput design of small-molecule binding sites | Efficient sampling of cofactor interactions | Creating artificial metalloenzymes with abiotic cofactors |
| RFdiffusion | Denoising diffusion probabilistic model | De novo backbone generation conditioned on functional motifs | High diversity of novel folds, user-specified constraints | Generating entirely novel scaffolds from minimal functional inputs |
| ProteinMPNN | Neural network-based inverse folding | Sequence design for a fixed protein backbone | Extreme speed and high accuracy in sequence design | Final sequence design for any de novo generated backbone |
Integrated Workflow forDe NovoEnzyme Design
The following diagram illustrates a generalized, iterative workflow for designing a functional enzyme using computational scaffolding, integrating the tools discussed above.
De Novo Enzyme Design and Optimization Workflow
Case Study: Designing an Artificial Metathase
A recent breakthrough provides a concrete example of this workflow, combining RifDock and Rosetta to create a functional ArM [5].
- Step 1: Define Target Reaction & Theozyme: The goal was to perform ring-closing metathesis (RCM) in living cells. A synthetic Hoveyda-Grubbs catalyst derivative (Ru1) was designed with a polar sulfamide group to guide computational design.
- Step 2: Scaffold Generation & Motif Placement: The RifDock suite was used to enumerate interacting rotamers around Ru1 and dock them into a de novo-designed closed alpha-helical toroidal repeat protein (dnTRP) scaffold.
- Step 3: Sequence Design: Rosetta's FastDesign was used to optimize the protein sequence for binding, refining hydrophobic contacts and stabilizing H-bonding residues. From 21 initial designs, dnTRP_18 was identified as the most promising.
- Step 4: Computational Validation & Optimization: Binding affinity was measured (KD = 1.95 µM), and a point mutation (F116W) was introduced to improve it further (KD = 0.16 µM). This variant, dnTRP_R0, formed a stable 1:1 complex with the cofactor.
- Step 5: Experimental Testing & Iteration: The initial design showed activity. To boost performance, directed evolution was applied, creating variants with a ≥12-fold improvement in turnover number (TON ≥ 1,000), demonstrating excellent catalytic performance in E. coli cytoplasm.
Experimental Validation and Optimization Protocols
Computationally designed enzymes must be rigorously validated experimentally. The following protocols are standard in the field.
1In SilicoValidation
Before moving to the lab, designs are filtered computationally.
- Structure Prediction: Tools like AlphaFold 2/3 or ESMFold are used to predict the structure of the designed sequence de novo. A successful design typically shows a high confidence score (pLDDT/pAE) and a low root-mean-square deviation (RMSD) from the design model [33] [19].
- Molecular Dynamics (MD) Simulations: MD can assess the stability of the designed scaffold and active site under simulated physiological conditions, identifying potential flexible or unstable regions [3].
- Stability Prediction: Tools like DeepDDG can predict the change in stability (ΔΔG) caused by point mutations, helping to select the most stable variants for testing [3].
2In VitroandIn VivoCharacterization
- Expression and Purification: Designs are cloned, expressed in a system like E. coli, and purified via affinity chromatography (e.g., His-tag). Soluble expression is a primary indicator of successful folding [5] [8].
- Biophysical Characterization:
- Thermostability: Analyzed by measuring the melting temperature (Tm) using techniques like differential scanning fluorimetry (DSF) [5] [8].
- Binding Affinity: For ArMs, techniques like tryptophan fluorescence quenching or native mass spectrometry confirm cofactor binding and determine dissociation constants (KD) [5].
- Functional Assays: Catalytic activity is measured under relevant conditions. For the artificial metathase, activity was quantified by monitoring RCM product formation and calculating the turnover number (TON) [5].
Optimization Strategies
Initial designs often require optimization.
- Directed Evolution: This powerful method involves creating mutant libraries (e.g., via error-prone PCR) and screening for improved activity or stability. It was crucial for enhancing the artificial metathase's performance [5].
- Rational Re-design: Based on experimental structures (e.g., from X-ray crystallography), problematic regions can be identified and re-designed. In one study, a "disorganized loop" was replaced using a loop searching algorithm, resulting in enzymes with high activity and excellent stereoselectivity [31].
Table 2: Key Experimental Metrics for Validating De Novo Enzymes
| Validation Stage | Key Metric | Method/Tool | Interpretation of Success |
|---|---|---|---|
| Computational | Structural Accuracy | AlphaFold 2/3, ESMFold | High pLDDT, Low RMSD to design model |
| Active Site Geometry | Molecular Dynamics (MD) | Stable positioning of catalytic residues | |
| Folding Stability | DeepDDG, Rosetta Energy | Negative ΔΔG (stabilizing mutation) | |
| Biophysical | Protein Folding & Solubility | SDS-PAGE, Size-Exclusion Chromatography | High yield of soluble protein |
| Thermostability | Melting Temperature (Tₘ) | High Tₘ (e.g., >65°C) | |
| Cofactor Binding | Fluorescence Quenching, Native MS | Low KD (nM - µM range), 1:1 stoichiometry | |
| Functional | Catalytic Efficiency | Turnover Number (kcat), Specificity (kcat/Km) | High TON and catalytic efficiency |
| Stereoselectivity | Enantiomeric Excess (e.e.) | High e.e. for asymmetric synthesis | |
| In Vivo Function | Whole-Cell Biocatalysis | Significant product formation in cells |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Resources for De Novo Scaffolding
| Resource | Function/Role | Specific Examples & Notes |
|---|---|---|
| Software Suites | Core algorithms for structure prediction, design, and simulation. | Rosetta, Schrodinger Suite, MOE [3] [5] |
| Generative AI Models | De novo backbone generation and inverse sequence design. | RFdiffusion [19], ProteinMPNN [19], AlphaFold 3 [33] |
| Specialized Docking | Designing protein scaffolds for small-molecule cofactor binding. | RifDock [5] |
| Quantum Chemistry Software | Constructing theozymes by modeling transition states and optimizing catalytic geometry. | Gaussian, ORCA [22] |
| Databases | Source of natural protein structures and sequences for model training and fragment sourcing. | Protein Data Bank (PDB) [32], UniProt [3] |
| Expression Systems | Producing the designed protein for experimental validation. | E. coli (common), yeast, cell-free systems [5] [8] |
| Cofactors | Synthetic metal complexes for creating artificial metalloenzymes (ArMs). | Hoveyda-Grubbs catalyst derivatives (e.g., Ru1) [5] |
Computational scaffolding with Rosetta, RifDock, and AI-driven generative models has transformed the paradigm of enzyme design. These tools provide a direct path from a desired chemical reaction to a functional, de novo protein catalyst, as evidenced by the successful creation of enzymes for carbon–silicon bond formation and olefin metathesis in living cells [31] [5]. The integration of precise physical modeling with data-driven deep learning is overcoming historical challenges in active-site pre-organization and conformational dynamics.
The future of the field lies in the tighter integration of these tools into end-to-end workflows and in expanding their capabilities to design for complex functions like allostery and sophisticated multi-step catalysis. As these methods mature, the ability to design robust, efficient, and bespoke enzymes on demand will unlock new possibilities in green chemistry, drug development, and synthetic biology, fully realizing the potential of de novo enzyme design.
The quest to design enzymes with novel or enhanced functions is a central challenge in biotechnology, with profound implications for drug development, sustainable chemistry, and fundamental biological research. Traditional enzyme engineering methods, such as rational design and directed evolution, have achieved significant milestones but often operate within constrained regions of sequence space. The emergence of sophisticated machine learning (ML) methodologies is now fundamentally reshaping this landscape, enabling the computational creation of biocatalysts with tailored functions. This whitepaper details the core algorithmic advances—specifically contrastive learning and graph neural networks (GNNs)—powering a new generation of tools like CLIPzyme and EnzymeCAGE. These tools are shifting the paradigm from local optimization of existing enzymes to the global exploration of sequence-structure-function space, thereby accelerating the de novo design of novel enzyme functions.
At the heart of this revolution is the ability to model the complex relationship between an enzyme's architecture and its catalytic activity. Unlike traditional approaches that rely heavily on sequence homology, these ML-based methods learn underlying biophysical principles from data, allowing them to generalize to unseen reactions and protein folds. This capability is critical for advancing de novo enzyme design, where the goal is to create entirely new enzymes for non-natural or orphan reactions, moving beyond the limitations of naturally evolved scaffolds [3] [34].
Core Machine Learning Architectures in Enzyme Informatics
Contrastive Learning for Multi-Modal Data Alignment
Contrastive learning has emerged as a powerful paradigm for integrating information from disparate biological data modalities. Inspired by successful models in computer vision like CLIP (Contrastive Language–Image Pre-training), this approach is being adapted to align representations of enzyme structures and chemical reactions [34].
The fundamental objective is to learn a shared embedding space where representations of enzymes and the reactions they catalyze are positioned close together, while non-catalytic pairs are pushed apart. This is achieved through a contrastive loss function that operates on pairs of data. For a batch of N (reaction, enzyme) pairs, the similarity score ( s{ij} ) between reaction embedding ( ri ) and enzyme embedding ( pj ) is typically computed as the cosine similarity: [ s{ij} = \frac{ri}{\lVert ri \rVert} \cdot \frac{pj}{\lVert pj \rVert} ] The loss function then maximizes the similarity for positive pairs (i=j) while minimizing it for negative pairs (i≠j) within the batch [35]. This training paradigm enables the model to capture functional relationships without explicit manual labeling of what makes an enzyme suitable for a specific reaction, learning a data-driven "functional similarity" metric directly from the data [34].
Graph Neural Networks for Geometric Reasoning
Graph Neural Networks (GNNs) provide a natural framework for modeling the intricate 3D structures of enzymes and molecules. Unlike sequence-based models, GNNs operate on graph structures where nodes represent atoms or residues, and edges represent bonds or spatial proximities [36].
The key innovation in modern enzyme informatics is the use of SE(3)-equivariant GNNs, which respect the geometric symmetries of 3D space (rotation and translation). This means that rotating the input structure rotates the internal representations accordingly, ensuring predictions are geometrically consistent. These architectures are particularly adept at capturing the physical constraints of enzyme active sites and reaction mechanisms, as they can explicitly reason about atomic distances, angles, and torsions [36]. When processing a graph, GNNs perform message-passing operations where nodes aggregate information from their neighbors, allowing them to learn complex local environments that are critical for catalytic function prediction.
Cross-Attention Mechanisms
Cross-attention mechanisms enable models to learn which parts of an enzyme structure are most relevant to specific aspects of a chemical reaction, and vice versa. This is achieved by computing attention weights between all enzyme and reaction representation elements, allowing the model to focus on the most salient features for predicting functional relationships [36]. For enzyme design, this means the model can learn to associate specific active site residues with particular reaction centers—the atoms undergoing bond changes—without explicit supervision. This capability for explicit interaction modeling provides both performance gains and improved interpretability, as the attention weights can reveal potential catalytic mechanisms.
Tool-Specific Implementations and Methodologies
CLIPzyme: Reaction-Conditioned Virtual Screening
CLIPzyme implements a contrastive learning framework specifically designed for virtual enzyme screening, framing the challenge as a retrieval task where the goal is to rank enzymes according to their predicted catalytic activity for a query reaction [35] [34].
Table 1: CLIPzyme Architecture Components
| Component | Implementation | Key Innovation |
|---|---|---|
| Reaction Encoder | Processes substrate and product structures, simulating a pseudo-transition state from bond changes [35] | Moves beyond deterministic featurization to learn transition state features directly from data |
| Protein Encoder | Encodes AlphaFold-predicted structures to leverage 3D organization of conserved domains [35] | Enables precomputation of enzyme embeddings for efficient large-scale screening |
| Training Objective | Contrastive loss aligning enzyme and reaction representations in shared space [35] | Creates a functional similarity metric without relying on EC number classifications |
| Screening Approach | Cosine similarity between reaction and precomputed enzyme embeddings [34] | Allows rapid identification of candidate enzymes from large databases |
A critical innovation in CLIPzyme is its reaction encoding scheme, which models molecular structures of both substrates and products to simulate a pseudo-transition state based on the bond changes of the reaction. This approach aims to capture information about the transition state stabilization that is fundamental to enzymatic catalysis [35]. In evaluations, CLIPzyme achieved a BEDROC₈₅ of 44.69% in virtual screening scenarios where limited information on the reaction was available, outperforming Enzyme Commission (EC) number prediction baselines. Furthermore, combining CLIPzyme with EC predictors consistently yielded improved results, suggesting these approaches capture complementary aspects of enzyme function [35].
Figure 1: CLIPzyme's contrastive learning workflow for virtual enzyme screening.
EnzymeCAGE: Catalytic-Aware Geometric Learning
EnzymeCAGE (CAtalytic-aware GEometric-enhanced enzyme retrieval model) represents a more recent advancement that explicitly incorporates detailed geometric information about enzyme active sites and reaction centers through a multi-modal architecture [34].
Table 2: EnzymeCAGE Architecture Components
| Module | Implementation | Function |
|---|---|---|
| Geometry-Enhanced Pocket Attention | GNN with attention biased by inter-residue distances and dihedral angles [34] | Identifies catalytically important residues and their spatial relationships |
| Center-Aware Reaction Interaction | Attention mechanism focusing on reaction center atoms [34] | Captures dynamics of substrate-to-product conversion |
| Global Context Integration | ESM2 protein language model embeddings [34] | Provides evolutionary and sequence-level context |
| Multi-Modal Fusion | Combines pocket, reaction, and global features [34] | Enables comprehensive compatibility assessment |
The geometry-enhanced pocket attention module uses fine-grained structural information—such as inter-residue distances and dihedral angles—as an attention bias within a self-attention mechanism. This allows the model to prioritize catalytically important residues and understand their spatial relationships more accurately [34]. Simultaneously, the center-aware reaction interaction module assigns higher attention weights to atoms involved in bond changes during the chemical transformation. EnzymeCAGE demonstrated a 44% improvement in function prediction and a 73% increase in enzyme retrieval accuracy compared to traditional methods like BLASTp on the Loyal-1968 test set, achieving a Top-1 success rate of 33.7% and Top-10 success rate exceeding 63% [34].
EZSpecificity: SE(3)-Equivariant Substrate Prediction
EZSpecificity employs a cross-attention empowered SE(3)-equivariant GNN architecture specifically designed for predicting enzyme substrate specificity [36]. Trained on a comprehensive database of enzyme-substrate interactions, the model demonstrated remarkable accuracy in identifying reactive substrates, achieving 91.7% accuracy in experimental validation with eight halogenases and 78 substrates, significantly outperforming previous state-of-the-art models (58.3%) [36]. This approach is particularly valuable for characterizing enzyme promiscuity and identifying non-canonical substrates for biocatalytic applications.
Comparative Analysis of ML Approaches
Table 3: Performance Comparison of Enzyme Design Tools
| Tool | Core Methodology | Primary Application | Reported Performance | Key Advantage |
|---|---|---|---|---|
| CLIPzyme [35] [34] | Contrastive learning with reaction and enzyme encoders | Virtual screening of enzymes for novel reactions | BEDROC₈₅ = 44.69% | Effective with limited reaction information |
| EnzymeCAGE [34] | Geometric deep learning with pocket attention | Enzyme retrieval and function prediction | Top-1: 33.7%, Top-10: >63% | Interpretable through attention mechanisms |
| EZSpecificity [36] | SE(3)-equivariant GNN with cross-attention | Substrate specificity prediction | 91.7% accuracy on halogenases | High accuracy on challenging specificity problems |
| Squidly [37] | Contrastive learning on PLM embeddings | Catalytic residue prediction from sequence | F1=0.64 at <30% sequence identity | Sequence-only approach, 50x faster than folding |
Experimental Protocols and Validation Frameworks
Benchmark Datasets and Evaluation Metrics
Rigorous evaluation of enzyme design tools requires carefully constructed benchmarks that test generalizability rather than memorization. Key datasets used in the field include:
- Uni14230 and Uni3750: Curated from UniProt and M-CSA databases, filtered to <60% sequence identity to reduce redundancy [37].
- CataloDB: A recently introduced benchmark designed to address shortcomings of previous datasets by ensuring <30% sequence and structural identity between training and test sets [37].
- Loyal-1968 test set: Used for evaluating generalization to unseen enzymes, with EnzymeCAGE showing 44% improvement in function prediction on this benchmark [34].
Standard evaluation metrics include:
- BEDROC (Boltzmann-Enhanced Discrimination of ROC): Emphasizes early retrieval performance, with the α=85 parameter placing importance on the first ~10,000 ranked enzymes, corresponding to reasonable experimental screening capacity [35].
- Top-K accuracy: Particularly relevant for retrieval tasks, indicating whether the correct enzyme or reaction appears in the top K recommendations [34].
- F1 score: Balance between precision and recall, especially important for catalytic residue prediction as used in Squidly evaluation [37].
Experimental Validation Case Studies
Successful experimental validation is the ultimate test of computational predictions. Notable examples include:
- EZSpecificity validation: Testing with eight halogenases and 78 substrates showed 91.7% accuracy in identifying the single potential reactive substrate, demonstrating practical utility in predicting substrate specificity [36].
- Semantic design with Evo: A genomic language model (Evo) was used to generate novel toxin-antitoxin systems, including a functional bacterial toxin (EvoRelE1) that exhibited strong growth inhibition (approximately 70% reduction in relative survival) despite limited sequence similarity to known toxins [38].
- Squidly catalytic residue prediction: The tool achieved an F1 score of 0.64 on sequences with less than 30% identity to training data, demonstrating generalization capability while being 50x faster than structure-based approaches [37].
Table 4: Key Research Reagents and Computational Tools
| Resource | Type | Function in Research | Access |
|---|---|---|---|
| AlphaFold DB [35] [34] | Protein structure database | Provides predicted 3D structures for enzymes without experimental structures | Publicly available |
| BRENDA [35] | Enzyme function database | Curated data on enzyme specificity, reactions, and kinetics | Publicly available |
| M-CSA [37] | Catalytic Site Atlas | Manually curated enzyme mechanism data for training and validation | Publicly available |
| ESM2 [34] | Protein Language Model | Provides evolutionary context and sequence representations | Open source |
| UniProt [37] | Protein sequence database | Comprehensive sequence data with functional annotations | Publicly available |
| EnzymeMap [35] | Reaction database | Curated biochemical reactions for training reaction encoders | Publicly available |
| PyTorch Geometric | ML library | Implementation of GNNs and graph learning algorithms | Open source |
| RDKit | Cheminformatics | Molecular representation and fingerprint generation | Open source |
Figure 2: Ecosystem of tools, data sources, and models in ML-driven enzyme design.
Integration with De Novo Enzyme Design Pipelines
The ultimate goal of these predictive tools is their integration into end-to-end de novo enzyme design pipelines. While CLIPzyme and EnzymeCAGE excel at identifying natural enzymes that can be repurposed or optimized, they also provide critical components for fully de novo approaches [3].
The transition from prediction to generation involves using these models to guide the design of entirely new enzyme scaffolds. For instance, the functional insights gained from catalytic residue predictors like Squidly can inform the construction of theoretical catalytic sites (theozymes) that serve as blueprints for de novo design [37]. Furthermore, the reaction and enzyme representations learned by these models can condition generative algorithms to produce novel sequences with desired catalytic properties [3].
Semantic design approaches, as demonstrated with the Evo genomic language model, show how functional context can guide the generation of novel protein sequences. By prompting the model with sequences of known function, researchers can generate novel genes whose functions mirror those found in similar natural contexts, accessing new regions of functional sequence space [38]. This represents a shift from traditional biological design—which involves combining or optimizing characterized sequences—toward true de novo generation based on functional semantics.
Future Directions and Challenges
Despite significant progress, several challenges remain in the application of machine learning to enzyme design. Low efficiency in design processes persists, with methods like the "Family Hallucination" strategy requiring extensive screening to yield a few active enzymes [3]. The precise orchestration of catalytic residues in three-dimensional space remains a complex challenge that current methods still struggle to fully capture.
Future advancements will likely come from several directions:
- Improved reaction representations that better capture transition state chemistry and steric constraints.
- Integration of dynamics to account for protein flexibility and conformational changes during catalysis.
- Multi-objective optimization considering not just activity but also stability, solubility, and expressibility.
- Uncertainty quantification to better assess prediction reliability and guide experimental prioritization.
As these technical challenges are addressed, ML-driven enzyme design is poised to expand applications in drug development, green chemistry, and the synthesis of complex molecules that are currently inaccessible through biological means [39]. The shift from traditional experiment-driven models to data-driven computationally intelligent systems is already underway, promising to unlock new frontiers in biocatalysis and synthetic biology.
Directed Evolution and High-Throughput Screening for Functional Optimization
Directed evolution is a powerful protein engineering methodology that harnesses the principles of Darwinian evolution—iterative cycles of genetic diversification and selection—in a laboratory setting to optimize proteins for human-defined applications. This approach has matured from a novel academic concept into a transformative biotechnology, recognized by the 2018 Nobel Prize in Chemistry awarded to Frances H. Arnold for its development [40]. The primary strategic advantage of directed evolution lies in its capacity to deliver robust solutions for enhanced stability, novel catalytic activity, or altered substrate specificity without requiring detailed a priori knowledge of a protein's three-dimensional structure or catalytic mechanism [40].
Unlike rational design approaches that rely on predictive understanding of sequence-structure-function relationships, directed evolution explores vast sequence landscapes through mutation and functional screening, frequently uncovering non-intuitive and highly effective solutions that would not have been predicted by computational models or human intuition [40]. This capability makes it particularly valuable for optimizing complex protein functions where mechanistic understanding remains incomplete. Today, this technology is routinely deployed across pharmaceutical, chemical, and agricultural industries to create enzymes and proteins with properties optimized for performance, stability, and cost-effectiveness [40].
Core Principles of Directed Evolution
The Directed Evolution Cycle
At its core, directed evolution functions as a two-part iterative engine that drives a protein population toward a desired functional goal. This process compresses geological timescales of natural evolution into weeks or months by intentionally accelerating mutation rates and applying user-defined selection pressure [40]. The iterative cycle consists of two fundamental steps executed sequentially:
- Generation of Genetic Diversity: Creating a library of protein variants through mutagenesis.
- Selection or Screening: Applying a high-throughput screen or selection to identify rare variants exhibiting improvement in the desired trait [40].
The genes encoding these improved variants are then isolated and used as the starting material for the next evolution round, allowing beneficial mutations to accumulate over successive generations. A critical distinction from natural evolution is that the selection pressure is decoupled from organismal fitness; the sole objective is optimizing a single, specific protein property defined by the experimenter [40].
Navigating Protein Fitness Landscapes
The directed evolution workflow is essentially an algorithm for navigating the immense and complex fitness landscapes that map protein sequence to function [40]. Fitness landscapes are more rugged and difficult to traverse when rich in epistatic (non-additive) effects of amino acid substitutions [41]. Epistasis is often observed between mutations in close structural proximity and is enriched at binding surfaces or enzyme active sites due to direct interactions between residues, substrates, and/or cofactors [41].
A typical directed evolution experiment begins with a parent gene encoding a protein with basal-level desired activity. This gene undergoes mutagenesis to create a diverse variant library. These variants are then expressed as proteins, and the population is challenged with a screen or selection identifying individuals with improved performance [40]. For example, improving enzyme thermostability might involve heating the library to a temperature that denatures the parent protein, then screening variants for remaining catalytic activity [40]. This iterative process repeats until desired performance targets are met or no further improvements are found.
Library Creation Methods
The creation of a diverse gene variant library is the foundational step defining explorable sequence space boundaries. The quality, size, and nature of this diversity directly constrain potential outcomes of the entire evolutionary campaign [40]. Several methods introduce genetic variation, each with distinct advantages, limitations, and inherent biases shaping evolutionary trajectories [40].
Table 1: Library Creation Methods in Directed Evolution
| Method | Key Principle | Advantages | Limitations | Typical Mutational Outcome |
|---|---|---|---|---|
| Error-Prone PCR (epPCR) [40] | Modified PCR with reduced polymerase fidelity introduces random mutations during gene amplification. | - Straightforward implementation- Requires no structural information- Broad exploration of sequence space | - Mutational bias (favors transitions over transversions)- Limited amino acid accessibility (~5-6 of 19 possible alternatives per position)- Primarily generates point mutations | 1-2 amino acid substitutions per protein variant |
| DNA Shuffling [40] | Random gene fragmentation followed by recombination of homologous fragments. | - Mimics natural recombination- Combines beneficial mutations from multiple parents- Accelerates functional improvement | - Requires sequence homology (≥70-75% identity)- Non-uniform crossover distribution | Novel combinations of existing mutations; chimeric genes |
| Site-Saturation Mutagenesis [40] | Targeted mutagenesis to comprehensively explore all possible amino acids at specific residue positions. | - Deep, unbiased interrogation of specific residues- Higher quality, smaller libraries- Ideal for optimizing "hotspot" positions | - Requires prior knowledge (e.g., structural data, hotspot identification)- Limited to predefined residues | All 19 possible amino acids at targeted positions |
Strategic Implementation of Diversification Methods
The choice of diversification strategy is a critical decision shaping the entire evolutionary search. Relying on a single method can lead experiments into evolutionary dead ends due to inherent methodological biases [40]. A robust R&D strategy often employs methods sequentially [40]:
- Initial Exploration: epPCR identifies beneficial mutations from broad sequence space exploration.
- Recombination: DNA shuffling combines beneficial mutations from multiple parents.
- Targeted Optimization: Saturation mutagenesis exhaustively explores key hotspots identified in initial stages.
This combined approach ensures the most thorough exploration of promising fitness landscape regions [40]. For problems with known structural constraints or epistatic interactions, such as active site optimization, starting with saturation mutagenesis at key positions may be more efficient [42].
High-Throughput Screening Methodologies
The central challenge after creating a diverse variant library is identifying rare improved variants from a population dominated by neutral or non-functional mutants. This genotype-to-phenotype linking is the primary bottleneck in directed evolution [40]. Success follows the axiom: "you get what you screen for" [40]. The screening platform's power and throughput must match the library's size and complexity [40].
A key distinction exists between screening and selection [40]:
- Screening: Individual evaluation of every library member for the desired property.
- Selection: Establishing a system where desired function directly couples to host organism survival or replication, automatically eliminating non-functional variants.
Table 2: High-Throughput Screening and Selection Platforms
| Method Type | Specific Platform | Throughput | Key Advantages | Primary Limitations |
|---|---|---|---|---|
| Selection [40] | Survival-based coupling | Very high (10^7-10^12 variants) | - Automates identification- Handles extremely large libraries- Minimal labor intensive | - Difficult to design- Prone to artifacts- Provides limited activity distribution data |
| Colony Screening [40] | Agar plate assays | Medium (10^3-10^4 variants) | - Simple, established methodology- Visible phenotype (e.g., halo formation) | - Limited quantitative data- Lower throughput- Requires scalable assay |
| Microtiter Plate Screening [40] | 96- or 384-well formats | Medium (10^3-10^4 variants) | - Quantitative data collection- Compatible with automated liquid handling- Robust and reproducible | - Throughput limited by well number- Requires assay miniaturization |
| Robot-Assisted Screening [43] | Automated liquid handling | High (100s-1000s of proteins weekly) | - High reproducibility- Minimal human error- Reduced material cost and waste | - Initial equipment investment- Protocol development required |
Advanced Screening Implementation
Recent advances address traditional screening limitations. For example, a low-cost, robot-assisted pipeline enables purification of 96 proteins in parallel with minimal waste, scalable for processing hundreds of proteins weekly per user [43]. This platform uses affordable liquid-handling robots (e.g., Opentrons OT-2) and small-scale E. coli expression to achieve sufficient yields for comprehensive thermostability and activity analyses [43].
Statistical experimental design is crucial in developing and optimizing high-throughput screening assays where numerous variables and potential interactions exist [44]. These methods help efficiently identify optimal assay conditions for robotic implementation [44].
Machine Learning-Enhanced Directed Evolution
Overcoming Epistasis with Machine Learning
Conventional directed evolution faces limitations when mutations exhibit non-additive (epistatic) behavior, potentially becoming trapped at local optima on rugged fitness landscapes [42]. Machine learning (ML) techniques circumvent these obstacles by providing strategies to navigate complex landscapes more efficiently [42].
Various ML-assisted directed evolution (MLDE) strategies identify high-fitness protein variants more efficiently than typical directed evolution approaches [41]. MLDE utilizes supervised ML models trained on sequence-fitness data to capture non-additive effects, enabling prediction of high-fitness variants across the entire landscape [41].
Active Learning-Assisted Directed Evolution
Active Learning-assisted Directed Evolution (ALDE) represents an advanced MLDE approach employing iterative machine learning with uncertainty quantification to explore protein search space more efficiently than current methods [42]. ALDE alternates between collecting sequence-fitness data through wet-lab experimentation and computationally training ML models to prioritize new sequences for screening [42].
In application to an challenging engineering landscape—optimizing five epistatic residues in a protoglobin active site—ALDE improved yield of a desired cyclopropanation product from 12% to 93% in just three rounds, exploring only ~0.01% of the design space [42]. The final variant contained mutations not expected from initial single-mutation screens, demonstrating that epistasis consideration through ML-based modeling is crucial [42].
ALDE Workflow: Machine learning-guided directed evolution cycle.
Performance Across Diverse Landscapes
Comprehensive analysis across 16 diverse combinatorial protein fitness landscapes demonstrates that MLDE strategies generally exceed or match directed evolution performance [41]. Advantages become more pronounced as landscape attributes pose greater obstacles for directed evolution (e.g., fewer active variants and more local optima) [41].
Focused training using zero-shot predictors—which leverage evolutionary, structural, and stability knowledge without experimental data—further improves MLDE performance by enriching training sets with more informative variants [41]. This approach consistently outperforms random sampling for both binding interactions and enzyme activities [41].
Experimental Protocol: ALDE for Enzyme Optimization
This protocol adapts the ALDE methodology successfully used to optimize a protoglobin for non-native cyclopropanation activity [42].
Initial Library Construction
- Define Design Space: Select k target residues (typically 3-5 positions) based on structural knowledge or prior mutagenesis data. For enzyme active sites, include residues in close structural proximity with evidence of epistatic interactions [42].
- Library Synthesis: Create an initial variant library by simultaneously mutating all k positions using sequential PCR-based mutagenesis with NNK degenerate codons [42].
- Initial Screening: Express and screen library variants using a functional assay. For the protoglobin case, screening was performed via gas chromatography for cyclopropanation products [42].
Machine Learning-Guided Rounds
- Model Training: Use initial sequence-fitness data to train a supervised ML model. The ALDE codebase (https://github.com/jsunn-y/ALDE) supports various sequence encodings and models with uncertainty quantification [42].
- Variant Prioritization: Apply an acquisition function to the trained model to rank all design space sequences. Balance exploration and exploitation using appropriate acquisition functions [42].
- Iterative Experimentation: Test top N predicted variants (typically tens to hundreds) in the wet lab. Collect new sequence-fitness data and repeat cycles until fitness is sufficiently optimized [42].
Key Reagents and Equipment
Table 3: Essential Research Reagents and Equipment for Directed Evolution
| Category | Item | Specification/Function |
|---|---|---|
| Library Construction | Polymerase Chain Reaction (PCR) reagents | Standard and error-prone PCR protocols |
| NNK degenerate codons | Allows all amino acids at targeted positions | |
| Competent E. coli cells | Zymo Mix & Go! or equivalent for transformation [43] | |
| Screening & Expression | Expression vector | pCDB179 or equivalent with affinity tag (e.g., His-tag) [43] |
| Affinity purification resin | Ni-NTA magnetic beads or equivalent [43] | |
| Liquid handling robot | Opentrons OT-2 or equivalent for automation [43] | |
| Deep-well plates | 24-well format for expression cultures [43] | |
| Analysis | Functional assay reagents | Substrate- and product-specific detection |
| Chromatography system | GC or HPLC for product separation and quantification [42] |
Integration with De Novo Enzyme Design
Directed evolution and de novo enzyme design represent complementary approaches in the protein engineering toolkit. While directed evolution optimizes existing proteins, de novo design aims to create entirely novel enzymes from scratch. Recent advances in deep learning methods like RFdiffusion enable design of diverse functional proteins from simple molecular specifications [19].
Mechanistic rules for de novo design are emerging, providing principles for engineering systems that give direction to chemistry [45]. These rules—including friction matching between enzyme and substrate, comparable conformational changes, and appropriate timing—can provide valuable input for machine learning algorithms in directed evolution [45].
The integration of machine learning across both domains creates powerful synergies. As demonstrated by RFdiffusion, deep-learning frameworks can solve diverse design challenges, including de novo binder design and symmetric architecture creation [19]. Similarly, ML-assisted directed evolution methods like ALDE efficiently navigate complex fitness landscapes where epistasis presents significant challenges [42].
Future Perspectives
The convergence of directed evolution with machine learning and de novo design represents the future of protein engineering. Computational studies confirm that ML-assisted directed evolution offers significant advantages across diverse protein fitness landscapes, particularly those challenging for conventional directed evolution [41].
Future developments will likely focus on:
- Improved uncertainty quantification in ML models for better exploration-exploitation balance [42]
- Integration of zero-shot predictors leveraging evolutionary, structural, and stability information [41]
- Development of more automated and miniaturized screening platforms [43]
- Enhanced methods for tackling higher-dimensional design spaces [42]
As these technologies mature, the boundary between optimizing natural proteins and creating entirely novel enzymes will continue to blur, enabling unprecedented control over protein function for therapeutic, industrial, and research applications.
The integration of synthetic metal complexes into biological systems represents a frontier in expanding the functional capabilities of proteins. Cofactor engineering aims to design artificial metalloenzymes that perform novel transformations not found in nature while operating efficiently within cellular environments. This field sits at the intersection of bioinorganic chemistry, computational biology, and synthetic biology, offering pathways to address challenges in biocatalysis, bioremediation, and therapeutic development. The fundamental challenge lies in overcoming nature's own constraints—particularly the Irving-Williams series that governs metal affinity in biological systems—to create functional complexes that can be predictably integrated into proteins and cells [46].
Within the broader context of de novo enzyme design, cofactor engineering provides a critical bridge between abiotic catalysis and biological compatibility. While natural metalloenzymes have evolved exquisite metal specificity and catalytic efficiency, their repertoire is limited to biologically relevant reactions and conditions. The strategic redesign of metal-binding sites or creation of entirely new metallopeptides enables access to non-biological chemistry while maintaining the selectivity and green chemistry advantages of enzymatic catalysis [47]. This technical guide examines the computational and experimental methodologies enabling this emerging capability, with particular emphasis on overcoming the persistent challenge of mismatched metal availability in heterologous expression systems [48] [46].
Fundamental Principles of Biological Metal Recognition
The Irving-Williams Series and Its Engineering Implications
A foundational concept in cofactor engineering is the Irving-Williams series, which describes the inherent stability trend for divalent metal complexes in biological systems: Mn(II) < Fe(II) < Co(II) < Ni(II) < Cu(II) > Zn(II). This thermodynamic preference presents a significant engineering challenge, as proteins often bind non-cognate metals that follow this series rather than their intended biological cofactors [46]. Experimental studies with the cyanobacterial Mn(II)-cupin MncA demonstrate that metal preferences during folding and trapping faithfully follow this series, with Cu(I) showing approximately 4 × 10⁷-fold preference over Mn(II) [46]. This creates a natural mis-metalation problem when expressing metalloproteins in heterologous systems where intracellular metal availabilities differ from native environments.
Understanding relative metal binding affinities is crucial for predicting and engineering metalation states. Recent research has enabled quantification of these preferences through refolding competitions in buffered metal solutions. The table below summarizes experimentally determined metal binding preferences for MncA relative to Mn(II) [46]:
Table 1: Metal binding preferences of MncA during folding relative to Mn(II)
| Metal | Relative Preference | Competition Method |
|---|---|---|
| Cu(I) | 4 × 10⁷-fold | Bicinchoninic acid (BCA) buffer |
| Cu(II) | 4 × 10⁴-fold | NTA buffer |
| Zn(II) | 1.4 × 10³-fold | NTA buffer |
| Ni(II) | 2.9 × 10²-fold | Histidine buffer |
| Co(II) | 3.8 × 10¹-fold | NTA buffer |
| Fe(II) | 2.0 × 10¹-fold | NTA buffer |
These quantitative preferences enable predictive modeling of metalation states when combined with knowledge of intracellular metal availability, forming the basis for rational design strategies.
Computational Framework for Cofactor Design
Structure-Based Design Approaches
Structure-based computational design has become indispensable for engineering metal-binding sites into proteins. The Rosetta software suite (version 3.14) provides a comprehensive platform for macromolecular modeling that employs physics-based energy functions to predict stable conformations from 3D constraints [49]. This approach has been successfully applied to design proteins with novel metalloenzyme activities, including the creation of porphyrin-containing proteins that serve as efficient and stereoselective catalysts [31]. The methodology typically begins with a blueprint of secondary structure elements, employing fragment assembly and force-field energy minimization to fold proteins in silico before selecting the lowest-energy conformations as candidate designs [50].
Complementing traditional physics-based approaches, machine learning integration has dramatically enhanced computational design capabilities. AlphaFold has achieved unprecedented accuracy in predicting protein structures from amino acid sequences, while RoseTTAFold offers robust performance in modeling protein complexes [49]. The synergy between data-driven machine learning and physics-based modeling enables more robust and reliable computational pipelines. For metalloprotein design, this integration is particularly valuable for predicting metal-binding sites and their coordination geometries, though challenges remain in accurately modeling the structural impacts of metal incorporation and point mutations near active sites [49].
Bioinformatics-Driven Minimalist Design
An alternative to structure-based design is the bioinformatics approach, which leverages evolutionary information to design minimal functional sites. The MetalSite-Analyzer (MeSA) tool exemplifies this strategy by enabling researchers to extract conserved sequence motifs for binding specific metals from natural protein databases [47]. This tool analyzes the minimal functional site (MFS)—the local three-dimensional environment including all residues within 5Å of any metal-binding ligand—to identify conserved residues critical for metal coordination and catalysis.
This approach has successfully designed H4pep, an eight-residue peptide (HTVHYHGH) that mimics the trinuclear copper site of laccase enzymes. Despite its simplified structure, Cu(II) binding to H4pep forms a Cu²⁺(H4pep)₂ complex with a β-sheet secondary structure that demonstrates catalytic activity for O₂ reduction [47]. This minimalist design strategy offers advantages in synthetic accessibility, stability under non-physiological conditions, and interfacial electron transfer capability due to smaller molecular cross-sections.
Table 2: Key computational tools for cofactor engineering
| Tool | Methodology | Application in Cofactor Engineering |
|---|---|---|
| Rosetta | Physics-based energy minimization | De novo protein design, metal-binding site design |
| AlphaFold | Deep learning structure prediction | Protein structure prediction, mutation impact analysis |
| RoseTTAFold | Deep learning with physical constraints | Protein complex modeling, conformational ensembles |
| MetalSite-Analyzer (MeSA) | Bioinformatics, sequence conservation analysis | Minimal functional site design, metal-binding motif identification |
| PROSS algorithm | Stability optimization | Designing soluble, stable metalloprotein variants |
Experimental Methodologies and Workflows
Integrated Design-Validation Workflow
A comprehensive workflow for cofactor engineering bridges computational design and experimental validation through iterative refinement. The following diagram illustrates this integrated approach:
Diagram Title: Integrated Workflow for Cofactor Engineering
This workflow has been successfully implemented in designing porphyrin-containing proteins as efficient and stereoselective catalysts. Initial computational designs based on simple helical bundle proteins were optimized through iterative redesign, where X-ray crystallography revealed structural discrepancies (e.g., disorganized loops instead of designed helices) that informed subsequent computational improvements [31]. This iterative process, combining AI-based protein design with chemical intuition and specialized algorithms, ultimately produced designs with high activity and excellent stereoselectivity.
Metalation Assessment Protocols
Determining metal binding specificity and affinity requires carefully controlled experimental conditions. For assessing metal preferences during protein folding, the following protocol has been established:
- Apo-protein Preparation: Express the target protein in E. coli to form inclusion bodies, then solubilize in urea to obtain unfolded apo-protein [46].
- Refolding Competition: Dilute urea-solubilized protein into urea-free buffer containing pairs of competing metals buffered with NTA (or histidine for Ni(II) competitions) [46].
- Anaerobic Handling: Perform competitions involving Fe(II) and Cu(I) in an anaerobic chamber with N₂-purged buffers, with metal stocks confirmed >95% reduced immediately before use [46].
- Protein-Metal Complex Separation: Recover refolded protein by anion exchange chromatography, then resolve from unbound metal by size exclusion chromatography (SEC) [46].
- Metal Analysis: Analyze SEC fractions (0.5 mL) for protein by UV absorbance and metals by ICP-MS to determine metal-protein stoichiometry [46].
This protocol enables quantitative measurement of metal preferences during the folding process, which is critical for predicting in vivo metalation states.
Solubility and Stability Enhancement
A common challenge in heterologous expression of metalloproteins is poor solubility and stability. Computational approaches like the Protein Repair One-Stop Shop (PROSS) algorithm address this by optimizing protein sequences to lower the free energy of the native state [48]. Applied to the electron donor protein AnfH of Fe-only nitrogenase, which was mostly insoluble when expressed in plant mitochondria, PROSS designed eight variants with improved soluble expression. The most successful variant (AnfH V6, containing T200A T228V E241H substitutions) showed approximately 90-fold greater abundance in the soluble fraction while maintaining functionality after [Fe₄S₄] cluster reconstitution [48].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of cofactor engineering requires specialized reagents and computational resources. The following table details key components of the experimental toolkit:
Table 3: Essential research reagents and materials for cofactor engineering
| Reagent/Material | Function/Application | Example Use Case |
|---|---|---|
| NTA (Nitrilotriacetic acid) | Metal buffering in competition assays | Maintaining defined metal concentrations during refolding studies [46] |
| Bicinchoninic acid (BCA) | Cu(I) buffering and detection | Cu(I) binding affinity measurements [46] |
| Size Exclusion Chromatography (SEC) columns | Separation of metal-bound protein from free metals | Purification of metalloproteins for stoichiometry analysis [46] |
| ICP-MS (Inductively Coupled Plasma Mass Spectrometry) | Quantitative metal analysis | Determining metal-protein stoichiometry [46] |
| CD (Circular Dichroism) Spectroscopy | Secondary structure determination | Verification of designed fold in metallopeptides [47] |
| PROSS Algorithm | Protein stability optimization | Designing soluble variants of metalloproteins [48] |
| MetalSite-Analyzer (MeSA) | Bioinformatics analysis of metal sites | Identifying conserved metal-binding motifs [47] |
| RFdiffusion | Generative protein design | Creating novel protein scaffolds for metal incorporation [49] |
Applications and Case Studies
Nitrogenase Engineering for Agricultural Applications
Cofactor engineering enables ambitious applications such as engineering nitrogenase directly into crops to reduce dependence on synthetic fertilizers. The Fe-only nitrogenase is a particularly promising target due to its simpler maturation pathway and lack of heterometal requirements [48]. However, the obligate electron donor protein AnfH from A. vinelandii proved mostly insoluble when expressed in plant mitochondria. Computational design using the PROSS algorithm and Rosetta energy calculations created eight AnfH variants with improved soluble expression, with the best variant (AnfH V6) showing approximately 90-fold greater abundance while maintaining functionality [48]. This demonstrates how computational design can overcome critical bottlenecks in complex metabolic engineering projects.
Biomimetic Peptide Catalysts
Short peptide scaffolds offer a minimalist approach to biomimetic catalyst design. The H4pep sequence (HTVHYHGH), designed using the MeSA bioinformatics tool to mimic the trinuclear copper site of laccase, demonstrates the potential of this approach [47]. Despite its minimal length, Cu(II) binding to H4pep forms complexes with β-sheet secondary structure that catalyze O₂ reduction. These metallopeptide complexes offer advantages including synthetic accessibility, stability across varied conditions, and efficient interfacial electron transfer—making them promising for applications in bioelectrocatalysis and sustainable energy conversion [47].
Future Perspectives and Challenges
As cofactor engineering advances, several challenges remain at the forefront of the field. Accurately predicting the structural and functional impacts of metal incorporation, especially for non-biological elements, requires improved computational methods that better account for metal-protein interactions [50]. The development of more sophisticated metalation calculators that incorporate intracellular metal availability and competition will enhance our ability to predict in vivo metalation states [46]. Additionally, expanding the repertoire of non-biological reactions catalyzed by designed metalloenzymes represents both a challenge and opportunity for the field.
The integration of cofactor engineering with de novo enzyme design promises to unlock new catalytic capabilities beyond nature's repertoire. As summarized by researchers in the field, "If people could design very efficient enzymes from scratch, you could solve many important problems" [31]. This potential is being realized through workflows that combine computational design with experimental validation, enabling the creation of enzymes that operate via mechanisms not previously known in nature [31]. As these methodologies mature, cofactor engineering will play an increasingly central role in expanding the functional universe of proteins for applications in medicine, energy, and sustainable manufacturing.
The field of drug development is undergoing a profound transformation, moving beyond the inhibition of single targets to sophisticated strategies that precisely control drug activity and target complex disease pathways. This evolution is particularly evident in the context of de novo design of novel enzyme functions, where computational and structural biology converge to create custom-tailored therapeutic agents. The integration of prodrug activation technologies with advanced understanding of disease pathway biology represents a frontier in precision medicine, enabling researchers to develop therapies with unprecedented specificity and reduced off-target effects. This whitepaper examines cutting-edge applications in drug development, focusing on three principal areas: photodynamic prodrug activation for oncology, computational design of protein-based inhibitors for antiviral applications, and innovative approaches to targeting key immune signaling pathways. These approaches demonstrate how modern drug development leverages multi-disciplinary strategies to address longstanding challenges in therapeutic efficacy and safety, providing a framework for researchers developing next-generation treatments for complex diseases.
Photodynamic Prodrug Activation in Oncology
Fundamental Mechanisms and Design Strategies
Photodynamic therapy (PDT) has emerged as a powerful platform for spatially and temporally controlled prodrug activation in cancer treatment. The core mechanism involves photosensitizers (PS) that generate reactive oxygen species (ROS) under specific light irradiation, which can subsequently trigger the release of active drug molecules from inert prodrug forms. This approach addresses a fundamental challenge in chemotherapy: the systemic toxicity and lack of specificity associated with traditional chemotherapeutic agents [51].
The design of light-activated prodrugs primarily follows two strategic pathways:
- Covalent Photosensitizer-Drug Conjugates: These systems incorporate a cleavable linker between the photosensitizer and the drug molecule. Upon light irradiation, ROS generated by the photosensitizer (typically singlet oxygen for Type II PDT) cleaves this linker, releasing the active drug payload.
- Non-covalent Nanocarrier Systems: These platforms co-encapsulate both photosensitizers and prodrugs within nanoparticle constructs. The ROS generated upon irradiation permeates the carrier matrix to activate the prodrug, providing enhanced control over release kinetics.
The critical advantage of photodynamic prodrug activation lies in its spatiotemporal precision. Unlike enzyme-activated prodrugs that may suffer from off-target activation due to enzyme presence in healthy tissues, light activation can be confined precisely to tumor regions, minimizing systemic exposure [51].
Experimental Protocol for Photodynamic Prodrug Evaluation
Materials and Equipment:
- Photosensitizer (e.g., porphyrin derivatives, AIEgens)
- Prodrug candidate with ROS-cleavable linker (e.g., amino acrylate, thioketal)
- Light source with appropriate wavelength filter
- Oxygen-controlled chamber
- ROS detection reagents (e.g., SOSG, DCFH-DA)
- HPLC system for drug release quantification
- Cell culture facilities
- Tumor cell lines
Methodology:
- Prodrug Synthesis: Conjugate therapeutic agent to ROS-cleavable linker. Common linkers include vinyl ethers, thioketals, or aryl boronate esters for Type I ROS.
- Formulation: Prepare nanoparticle encapsulation or conjugate with photosensitizer.
- In Vitro Activation Assay:
- Incubate prodrug system in PBS or cell culture medium
- Apply light irradiation at predetermined parameters (wavelength, intensity, duration)
- Monitor ROS generation using fluorescent probes
- Quantify drug release via HPLC at regular intervals
- Cellular Efficacy Testing:
- Treat tumor cells with prodrug system
- Apply localized light irradiation
- Assess cytotoxicity via MTT/WST assays
- Evaluate apoptosis/necrosis via flow cytometry
Key Parameters:
- Light wavelength matched to photosensitizer absorption
- Oxygen concentration (critical for Type II mechanisms)
- Prodrug:photosensitizer ratio
- Irradiation time and power density
Table 1: Comparison of Photodynamic Prodrug Activation Strategies
| Parameter | Covalent Conjugates | Non-covalent Nanocarriers |
|---|---|---|
| Drug Loading Capacity | Limited by conjugation sites | High (up to 50% w/w) |
| Release Kinetics | Typically faster | Tunable via carrier properties |
| Manufacturing Complexity | High (chemical synthesis) | Moderate (formulation) |
| Activation Efficiency | Dependent on linker chemistry | Dependent on ROS diffusion |
| Clinical Translation | Emerging | More established |
Computational Design of Miniprotein Inhibitors for Antiviral Applications
De Novo Design of SARS-CoV-2 Mpro Inhibitors
The de novo computational design of miniprotein inhibitors represents a groundbreaking application of novel enzyme function design in antiviral drug development. A prominent example is HB3-Core25, a computationally engineered miniprotein designed to disrupt the dimerization of SARS-CoV-2 Main Protease (Mpro), an essential enzyme for viral replication [52].
The design strategy leveraged the structural insight that the N-terminal region of Mpro (the "N finger") contributes approximately 39% of the homodimer interaction interface. Prior attempts using linear peptides mimicking the N-terminal sequence demonstrated proof-of-concept but suffered from limited potency (IC50 ≥ 500 μM) due to conformational flexibility. To address this, researchers employed Rosetta-based protein design to create a stable, trimeric helical bundle that effectively targets this flat protein-protein interface with significantly improved affinity (KD = 0.567 μM) and inhibitory activity [52].
Experimental Protocol for Computational Miniprotein Design
Computational Workflow:
- Structure Preparation: Obtain high-resolution crystal structure of target (PDB ID: 7ALI for Mpro). Remove crystallographic water molecules, ligands, and ions prior to energy minimization using Rosetta's FastRelax protocol.
- Binding Hotspot Identification: Perform alanine scanning using a consensus approach across FoldX, MutaBind2, and Rosetta's Flex ddG protocols to identify critical residues for binding.
- De Novo Miniprotein Design:
- Generate initial α-helical scaffold (H16) using RosettaRemodel
- Perform structural alignment of H16 Cα atoms with N8 hotspot residues
- Mutate valine residues at positions corresponding to R4 and M6 to arginine and methionine
- Execute backbone design using BluePrintBDR protocol with three-helix bundle topology
- Sequence Optimization: Conduct two-step sequence design (full monomer followed by interface optimization) using FastDesign protocol with 50,000 sequences.
- Validation: Assess designed structures using molecular dynamics simulations (GROMACS 2019.4 with CHARMM36m force field) to confirm stability.
Experimental Validation:
- In Vitro Production: Express and purify designed miniproteins
- Biophysical Characterization:
- Circular dichroism for secondary structure
- Thermal denaturation for stability assessment
- Surface plasmon resonance for binding affinity (KD)
- Functional Assays:
- Enzymatic activity inhibition (IC50 determination)
- Dimer disruption assays (size-exclusion chromatography)
Table 2: Key Research Reagent Solutions for Computational Protein Design
| Reagent/Resource | Function/Application | Source/Reference |
|---|---|---|
| Rosetta Software Suite | Protein structure prediction & design | [52] |
| FoldX | Protein stability & interaction calculations | [52] |
| GROMACS | Molecular dynamics simulations | [52] |
| CHARMM36m Force Field | Molecular dynamics parameters | [52] |
| EFI-EST Web Tool | Enzyme similarity network analysis | [53] |
| AlphaFold2 | Protein structure prediction | [52] |
| Pymol | Molecular visualization & manipulation | [52] |
Diagram 1: Computational Protein Design Workflow - This diagram illustrates the hierarchical process for de novo miniprotein design, from target identification through experimental validation.
Targeting Innate Immune Pathways: cGAS-STING Modulation
Allosteric Inhibition of cGAS for Autoimmune Applications
The cyclic GMP-AMP synthase (cGAS) - Stimulator of Interferon Genes (STING) pathway represents a crucial innate immune signaling axis with implications in autoimmunity, inflammation, and cancer. Recent advances in targeting this pathway demonstrate innovative approaches to allosteric inhibition and conditional activation. Researchers have developed protein condensation inhibitors (PCIs) that engage a novel allosteric site near the activation loop of cGAS, stabilizing it in a closed, inactive conformation that attenuates cGAS-DNA interactions [54].
The XL series inhibitors, particularly XL-3156 and XL-3158, exemplify structure-based drug design targeting this allosteric site. These compounds simultaneously occupy both allosteric and orthosteric sites, demonstrating cross-species potency and the ability to suppress cGAS-DNA condensate formation. This distinct mechanism triggers a morphological transition from liquid-solid phase separation to liquid-liquid phase separation at the molecular level, effectively modulating the phase behavior of cGAS [54].
Tumor-Specific STING Agonist Synthesis
Complementing the inhibition strategies for autoimmune applications, innovative approaches have emerged for tumor-specific STING activation. Researchers have developed a two-component prodrug system that enables the synthesis of a potent STING agonist specifically within tumor microenvironments [55].
This system leverages the unique mechanism of MSA-2, a non-cyclic dinucleotide STING agonist that forms non-covalent dimers before binding to STING. The approach utilizes two benign precursors: one bearing a caged nucleophile (activated by tumor-overexpressed enzymes like β-glucuronidase) and another containing an electrophile administered intratumorally. These components react through proximity-enhanced ligation to form a covalent, active dimer (SC2S) specifically within tumors, demonstrating submicromolar potency (EC50 = 0.71 μM) compared to the parent molecule (EC50 = 15 μM) [55].
Diagram 2: Two-Component STING Agonist System - This diagram illustrates the tumor-specific synthesis of a potent STING agonist from two benign precursors through enzyme activation and proximity-enhanced ligation.
Experimental Protocol for Two-Component Prodrug Evaluation
Materials:
- MSA-2 analogues with nucleophilic (thiol) and electrophilic (vinyl sulfonamide, chloroacetamide) modifications
- β-glucuronidase enzyme
- THP-1 Lucia ISG cell line (for STING pathway activation assessment)
- Purified human STING ligand-binding domain
- Isothermal titration calorimetry equipment
Methodology:
- Reactivity Assessment:
- Incubate nucleophilic and electrophilic components (50 μM each) in PBS, pH 7.0, 37°C
- Monitor reaction progression via HPLC/UV-Vis
- Compare rates to non-specific electrophiles as controls
Cellular Potency Determination:
- Treat THP-1 Lucia ISG cells with synthesized covalent dimers
- Measure interferon pathway activation using QUANTI-Luc assay
- Calculate EC50 values from dose-response curves
- Confirm pathway activation via Western blot for p-TBK1 and p-IRF3
Binding Affinity Measurement:
- Perform isothermal titration calorimetry with purified STING LBD
- Determine binding constants from thermogram data
In Vivo Tumor-Specific Activation:
- Administer nucleophilic component systemically in tumor-bearing mice
- Inject electrophilic component intratumorally
- Assess tumor-specific dimer formation via LC-MS
- Evaluate antitumor efficacy and immune cell infiltration
Table 3: Quantitative Data Comparison for STING Agonists
| Compound | Cellular EC50 (μM) | Binding Constant (nM) | Administration | Tumor Specificity |
|---|---|---|---|---|
| Endogenous 2',3'-cGAMP | ~0.001-0.01 | ~1-10 | Intratumoral | None |
| MSA-2 (Parent) | 15.0 | 18,000 (dimer KD) | Systemic | None |
| SC2S (Covalent Dimer) | 0.71 | 176 | Two-component system | High |
| CDN-based Agonists | 0.001-0.1 | ~1-10 | Intratumoral | None |
Regulatory Innovation for Advanced Therapies
The development of novel therapeutic modalities has prompted evolution in regulatory frameworks. The U.S. Food and Drug Administration has recently proposed a "Plausible Mechanism Pathway" designed to address the unique challenges of bespoke therapies, particularly for ultra-rare conditions where traditional randomized controlled trials are not feasible [56] [57].
This pathway centers on five core elements:
- Identification of a specific molecular or cellular abnormality
- Intervention targeting the underlying biological alteration
- Well-characterized natural history of the disease
- Confirmation of successful target engagement
- Demonstration of improvement in clinical outcomes
The framework leverages the expanded access single-patient IND paradigm as a foundation for marketing applications, with an emphasis on real-world evidence collection post-approval [56]. This regulatory innovation complements the scientific advances in drug development, potentially accelerating the translation of de novo designed therapies to clinical application.
Future Directions in Drug Development
The integration of prodrug activation strategies with targeted disease pathway modulation represents the future of precision therapeutics. The convergence of computational protein design, conditional activation technologies, and pathway-specific targeting enables researchers to develop therapies with unprecedented specificity. Emerging trends include the expanded application of PROteolysis TArgeting Chimeras (PROTACs), advances in radiopharmaceutical conjugates for precision oncology, and the continued evolution of CRISPR-based therapies for rare diseases [58].
For researchers in the field of de novo enzyme design, these developments highlight the importance of considering not only the structural and functional aspects of designed proteins but also their integration into broader therapeutic strategies that may include controlled activation, targeted delivery, and pathway-specific effects. The future of drug development lies in this multidisciplinary approach, combining deep biological insight with innovative engineering principles to create truly transformative medicines.
Navigating Design Challenges: Strategies for Enhancing Catalytic Efficiency and Biocompatibility
Overcoming Inefficient Preorganization and Transition State Stabilization
The de novo design of novel enzyme functions represents a frontier in biotechnology, with profound implications for therapeutic development, biocatalysis, and fundamental biological research. Central to this endeavor are two interconnected challenges: achieving efficient preorganization of the catalytic site and providing optimal transition state stabilization. Preorganization refers to the precise three-dimensional arrangement of catalytic residues and binding pockets that enables the enzyme to preferentially bind and stabilize the transition state of a chemical reaction, thereby dramatically lowering the activation energy [22]. In natural enzymes, evolutionary optimization has perfected these features; however, in de novo designed enzymes, inefficient preorganization and suboptimal transition state stabilization often result in catalytic efficiencies orders of magnitude below natural counterparts [22]. This technical guide examines the mechanistic underpinnings of these challenges and presents advanced computational and experimental methodologies for overcoming them, framed within the broader context of creating novel enzymatic functions for research and drug development.
Fundamental Challenges in De Novo Enzyme Design
The Preorganization Problem
Protein dynamics play a critical role in enzymatic catalysis, yet de novo designs often exhibit improper dynamic profiles that hinder function. Efficient enzymes balance structural rigidity with necessary flexibility—the catalytic site must be preorganized to recognize the transition state, but not so rigid as to prevent substrate binding or product release [59].
Nuclear magnetic resonance (NMR) studies on natural enzyme systems, such as FKBP12, reveal that successful catalysis often involves incremental rigidification upon binding. For instance, upon binding rapamycin, FKBP12 undergoes conformational selection where a subset of slow motions is quenched, preorganizing the protein for subsequent binding to mTOR [59]. This sequential rigidification enables precise molecular recognition. In de novo designs, this dynamic orchestration is frequently misaligned, leading to:
- Excessive flexibility in catalytic residues, reducing transition state stabilization
- Over-rigid structures that cannot accommodate the reaction coordinate
- Frustrated dynamics where the enzyme undergoes multiple non-productive conformational cycles
Recent research has identified three golden rules for optimal mechanochemical coupling in fueled enzymes: (1) enzyme and molecule should attach at the smaller end of each (friction matching), (2) conformational change of the enzyme must be comparable to or larger than that required of the molecule, and (3) the conformational change must be fast enough to actually stretch the molecule rather than just moving together [45].
Transition State Stabilization Deficits
Transition state stabilization is the cornerstone of enzymatic catalysis, yet remains exceptionally difficult to achieve in de novo designs. The fundamental principle, articulated by Linus Pauling and Richard Wolfenden, posits that efficient enzymes accelerate reactions by tightly binding and stabilizing the transition state [22]. Natural enzymes achieve transition state complementarity through precise electrostatic interactions, hydrogen bonding networks, and geometric constraints that have been evolutionarily optimized.
In de novo designs, transition state stabilization often fails due to:
- Imprecise electrostatic preorganization within active sites
- Suboptimal geometric positioning of catalytic residues
- Insufficient desolvation of the catalytic pocket
- Inadequate orbital overlap for covalent catalysis
Early de novo design efforts, particularly those utilizing RosettaMatch to place theozyme-derived catalytic motifs into existing protein scaffolds, typically produced catalysts with activities orders of magnitude below natural enzymes, primarily due to incomplete active-site preorganization and neglected conformational dynamics [22].
Table 1: Common Deficiencies in De Novo Enzyme Design and Their Consequences
| Deficiency Category | Specific Limitations | Impact on Catalytic Efficiency |
|---|---|---|
| Structural Preorganization | Improper backbone conformations around active site | Reduced transition state binding affinity |
| Inaccurate positioning of catalytic residues | Impaired chemical catalysis | |
| Suboptimal active site solvation/desolvation | Altered reaction energetics | |
| Dynamic Properties | Excessive microsecond-millisecond motions | Non-productive conformational sampling |
| Insufficient fast (ps-ns) dynamics | Impaired substrate access/product release | |
| Frustrated exchange cycles | Reduced turnover numbers | |
| Electrostatic Optimization | Inaccurate electrostatic potential shaping | Impaired transition state stabilization |
| Suboptimal protonation states | Altered pKa values of catalytic residues | |
| Poor charge distribution | Reduced rate enhancement |
Computational Strategies for Enhanced Preorganization
Generative AI for Backbone Design
The advent of generative artificial intelligence (GAI) has revolutionized de novo enzyme design by enabling the creation of novel protein scaffolds tailored to specific catalytic functions, rather than relying on repurposed natural scaffolds. RFdiffusion, a generative model based on the RoseTTAFold architecture, enables de novo construction of protein backbones with tailored topological features [19] [22]. By fine-tuning the structure prediction network on protein structure denoising tasks, RFdiffusion functions as a generative model that can:
- Create entirely novel protein folds not observed in nature
- Scaffold functional motifs with atomic accuracy
- Generate diverse solutions for a given design problem through stochastic denoising trajectories
Unlike earlier deterministic approaches that failed with minimalist active site descriptions, RFdiffusion builds structure progressively through many denoising iterations, requiring little starting structural information [19]. This capability was demonstrated in the design of a fully de novo serine hydrolase with catalytic efficiencies (kcat/Km) up to 2.2 × 10⁵ M⁻¹·s⁻¹ and folds distinct from natural hydrolases [22].
Diagram 1: RFdiffusion Backbone Generation Workflow
Active Site Identification Methods
Two complementary strategies have emerged for identifying and designing optimal active sites: data-driven consensus structure identification and first-principles theozyme design.
Consensus Structure Identification extracts conserved geometrical features from families of natural enzymes using structural databases like the Protein Data Bank. This approach identifies highly conserved spatial relationships and hydrogen-bonding networks associated with catalytic function [22]. For example, analysis of serine hydrolase families reveals not only the conserved catalytic triad (Ser-His-Asp) but also the precise geometry of the adjacent oxyanion hole—a microenvironment formed by backbone amide hydrogen atoms that stabilizes the tetrahedral intermediate [22].
Theoretical Enzyme Models (Theozymes) represent an "inside-out" strategy where an idealized minimal active site is constructed by placing key catalytic residues around a transition-state analogue [22]. This approach, pioneered by the Houk research group, involves:
- Transition State Characterization: Precisely locating the transition-state structure of the target reaction using quantum mechanical (QM) methods
- Catalytic Group Placement: Systematically positioning catalytic residue models (typically truncated side-chain fragments) around the transition state
- Geometry Optimization: Optimizing the arrangement to maximize transition state stabilization while minimizing reaction barrier
The hybrid functional B3LYP/6-31+G* remains one of the most widely applied methods for theozyme calculations, providing a favorable compromise between accuracy and efficiency with typical activation energy errors of approximately 1 kcal·mol⁻¹ [22].
Table 2: Computational Methods for Active Site Design
| Method Type | Key Features | Advantages | Limitations |
|---|---|---|---|
| Consensus Structure Identification | Statistical analysis of natural enzyme families | Leverages evolutionary solutions; Lower computational cost | Limited to reactions with natural templates; Does not explain why geometry is optimal |
| Theozyme Models | QM-based transition state optimization | First-principles approach; Applicable to novel reactions; Provides atomic-level insight | Computationally intensive; Requires expert knowledge |
| Machine Learning Approaches | Pattern recognition in sequence-structure-function space | High-throughput capability; Can identify non-obvious relationships | Dependent on training data quality; Limited mechanistic interpretability |
Diagram 2: Theozyme Construction Workflow
Advanced Methodologies for Transition State Stabilization
Multi-Timescale Dynamics Engineering
Protein dynamics across multiple timescales play crucial roles in enzymatic catalysis, yet traditional de novo design often neglected these dynamic considerations. NMR relaxation studies provide critical insights into the dynamic behavior of enzymes across picosecond-nanosecond (ps-ns) and microsecond-millisecond (μs-ms) timescales [59].
Research on FKBP12 demonstrates that binding events can affect fast fluctuations at regions distal to the binding interfaces, and that regions completely buried at high-affinity interfaces can still undergo μs-ms motions [59]. Perhaps counterintuitively, drug-bound enzymes retain the μs-ms motions critical to function in complexes with natural substrates, though with "frustrated" exchange cycles due to slow off-rates [59].
For de novo designs, engineering optimal dynamics involves:
- Rigidification of catalytic residues to maintain preorganization while allowing necessary flexibility for substrate binding and product release
- Preservation of functional μs-ms motions that enable catalytic cycling
- Minimization of non-productive frustrated dynamics that waste binding energy
Geometric Deep Learning for Functional Prediction
Recent machine learning approaches now enable more accurate prediction of enzyme function and compatibility with specific reactions, addressing the transition state stabilization challenge through improved virtual screening.
EnzymeCAGE (CAtalytic-aware GEometric-enhanced enzyme retrieval model) employs graph neural networks (GNNs) to create detailed local encodings of enzyme catalytic pockets and integrates these with global enzyme-level features from protein language models like ESM2 [34]. Key innovations include:
- Geometry-Enhanced Pocket Attention Module: Leverages fine-grained structural information (inter-residue distances, dihedral angles) as attention bias to pinpoint catalytically important residues
- Center-Aware Reaction Interaction Module: Focuses on reaction centers—specific atoms undergoing covalent bond changes—by assigning higher attention weights
- Catalytic-Specified Geometric-Enhanced Framework: Directly models enzyme-reaction compatibility rather than relying solely on EC number classifications
EnzymeCAGE demonstrated a 44% improvement in function prediction and 73% increase in enzyme retrieval accuracy compared to traditional methods like BLASTp and Selenzyme [34].
CLIPzyme adapts contrastive learning—successful in vision-language models—to align representations of enzyme structures and chemical reactions [34]. This framework:
- Trains separate encoders for enzyme structures and chemical reactions using a contrastive objective function
- Creates an embedding space where functionally related enzyme-reaction pairs have proximal representations
- Enables virtual screening by retrieving enzymes with embeddings closest to a query reaction's embedding
CLIPzyme particularly excels in scenarios with limited reaction information and shows strong generalization to unseen reactions and protein clusters [34].
Experimental Protocols and Validation
Quantum Mechanical Workflow for Theozyme Construction
Protocol Objective: Construct an accurate theozyme model for transition state stabilization of a target reaction.
Materials and Methods:
- Computational Environment: High-performance computing cluster with quantum chemistry software (Gaussian, ORCA, or similar)
- Method Selection: Hybrid functional (B3LYP) with 6-31+G* basis set for optimal accuracy/efficiency balance
- System Preparation:
- Construct transition state model of target reaction in gas phase
- Truncate catalytic residues to side-chain fragments, cap with hydrogens
- Position catalytic groups around transition state using crystallographic data or mechanistic knowledge
Procedure:
- Transition State Optimization:
- Perform conformational search to locate transition state structure
- Verify transition state with frequency calculation (exactly one imaginary frequency)
- Calculate intrinsic reaction coordinate to confirm connection to correct reactants and products
Catalytic System Assembly:
- Position catalytic residue fragments around transition state
- Freeze transition state coordinates during initial optimization
- Gradually release constraints during geometry optimization
Energy Evaluation:
- Calculate binding energy between catalytic groups and transition state
- Compare with natural enzyme systems where available
- Perform single-point energy calculations with larger basis sets if needed
Geometric Parameter Extraction:
- Measure critical distances (H-bond lengths, catalytic atom distances)
- Record angles and dihedrals defining catalytic geometry
- Compile parameters for scaffold design phase
Validation: Compare calculated activation energy barrier reduction with experimental data for similar reactions; target error < 1 kcal·mol⁻¹ [22].
Consensus Structure Identification Protocol
Protocol Objective: Identify conserved structural features in natural enzyme families for transfer to de novo designs.
Materials and Methods:
- Data Sources: Protein Data Bank, enzyme family databases (MEROPS, CAZy, etc.)
- Software Tools: Structural alignment tools (PyMOL, Chimera), sequence analysis packages
- Selection Criteria: Enzymes with experimentally verified activity, high-resolution structures (<2.0 Å), diverse phylogenetic origin
Procedure:
- Family Compilation:
- Select enzyme family based on target reaction
- Curate structures with common EC number or mechanistic similarity
- Ensure structural diversity to distinguish conserved vs. variable features
Structural Alignment:
- Align structures based on catalytic residues
- Superpose transition state analogues or inhibitors where available
- Identify structurally conserved regions beyond sequence homology
Geometric Analysis:
- Measure distances between catalytic atoms across family members
- Calculate angles and dihedrals defining active site geometry
- Identify conserved hydrogen bonding networks
Consensus Model Building:
- Extract most frequent values for key geometric parameters
- Build pseudo-protein representing family-average active site
- Identify structural outliers and correlate with functional variations
Validation: Check consensus model against catalytically competent natural enzymes; validate predictive power through mutant design and testing [22].
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools for De Novo Enzyme Design
| Category | Specific Tool/Reagent | Function/Application | Key Features |
|---|---|---|---|
| Generative AI Tools | RFdiffusion [19] [22] | De novo protein backbone generation | RoseTTAFold architecture; Conditional generation; High design success rates |
| ProteinMPNN [19] [22] | Protein sequence design | Fast sequence optimization; High recovery rates; Compatible with RFdiffusion | |
| Quantum Chemistry Software | Gaussian [22] | Theozyme construction and optimization | Extensive method library; B3LYP functional; Transition state optimization |
| ORCA [22] | Quantum mechanical calculations | Efficient for large systems; Multiple QM methods; Good performance/accuracy balance | |
| Machine Learning Frameworks | EnzymeCAGE [34] | Enzyme function prediction and retrieval | Geometric deep learning; Pocket attention mechanism; Multi-modal architecture |
| CLIPzyme [34] | Enzyme-reaction matching | Contrastive learning; Joint embedding space; Virtual screening | |
| Dynamics Characterization | NMR Spectroscopy [59] | Multi-timescale dynamics measurement | Atomic-resolution dynamics; Multiple timescales; Site-specific information |
| Molecular Dynamics [59] | Computational dynamics simulation | Atomic-level trajectory analysis; Timescale coverage; Mutational prediction | |
| Experimental Validation | Directed Evolution [60] [22] | Functional optimization of designs | Laboratory evolution; Activity screening; Functional improvement |
| Kinetic Analysis | Catalytic efficiency measurement | kcat/Km determination; Mechanistic insight; Comparison with natural enzymes |
Overcoming inefficient preorganization and transition state stabilization represents the central challenge in de novo enzyme design. The integration of generative AI for scaffold design, quantum mechanical methods for active site optimization, and geometric deep learning for function prediction has created a powerful toolkit for addressing these fundamental limitations. By explicitly considering multi-timescale dynamics, electrostatic preorganization, and transition state complementarity, next-generation de novo enzymes can achieve catalytic efficiencies approaching natural systems while performing novel chemical transformations. As these computational methodologies mature and integrate with high-throughput experimental validation, the prospect of designing bespoke enzymes for specific therapeutic and industrial applications becomes increasingly attainable, opening new frontiers in biotechnology and drug development.
Addressing Cofactor Instability and Inactivation in Complex Cellular Milieus
The de novo design of novel enzyme functions represents a frontier in synthetic biology, with profound implications for drug development, sustainable chemistry, and fundamental biological research. Within this paradigm, cofactor instability and inactivation emerge as critical bottlenecks that constrain the functional implementation of engineered enzymes in complex cellular environments. Cofactors—organic or inorganic molecules essential for enzymatic activity—serve as molecular switches that control diverse biological processes, but their functionality is notoriously susceptible to disruption in cellular milieus [61]. The transcription factor MYC, for instance, depends on dynamic cofactor interactions to regulate proliferation, apoptosis, and tumorigenesis, with its molecular and biological functions switching based on recruited cofactor complexes [61]. This inherent vulnerability extends to metabolic engineering, where cofactor imbalance significantly contributes to metabolic burden, diverting essential resources like NAD(P)H from host growth and native metabolism [62].
Understanding cofactor inactivation mechanisms is thus prerequisite to engineering robust enzyme systems. Research has identified several principal inactivation pathways: aggregation, thiol-disulfide exchange, alterations in primary structure, dissociation of cofactor molecules from enzyme active sites, subunit dissociation in oligomeric proteins, and conformational changes [63]. Often, conformational transformations trigger other inactivation mechanisms, creating cascading failure in enzymatic function. For enzyme engineers, these vulnerabilities represent both a challenge to overcome and a design parameter to address in creating functionally stable systems.
Molecular Mechanisms of Cofactor Instability
Principal Inactivation Pathways
The instability of enzyme-cofactor complexes arises from interrelated mechanisms that operate across temporal and spatial scales. At the molecular level, the dissociation of cofactor molecules from enzyme active sites represents a fundamental inactivation pathway [63]. This dissociation can be triggered by conformational changes in the protein structure, which may themselves result from perturbations in the cellular environment. The aldehyde dehydrogenase Cphy1178 from Clostridium phytofermentans, for instance, requires precise coordination of its CoA and NAD+ cofactors within distinct binding pockets to maintain catalytic function [64]. Structural analyses reveal that the adenine nucleotides of these cofactors adopt different conformations within the Rossman fold domain, creating a sophisticated coordination mechanism vulnerable to disruption [64].
Additional inactivation pathways include aggregation, where proteins form non-functional multimers; thiol-disulfide exchange, which disrupts crucial disulfide bonds; and alterations to the protein's primary structure [63]. These mechanisms frequently interweave, with conformational changes often triggering other inactivation processes. In microbial cell factories, metabolic toxicity from accumulated substrates, intermediates, or products induces oxidative stress that disrupts cellular architecture and inhibits essential protein activities, further compounding cofactor instability [62]. Reactive oxygen species generated through metabolic processes damage DNA, proteins, and lipids, creating a cascade of cellular dysfunction that ultimately impacts cofactor stability and function.
Environmental Stressors in Cellular Environments
In complex cellular environments, engineered enzymes face multiple stressors that accelerate cofactor inactivation. Fluctuations in pH, temperature, oxygen availability, and substrate concentrations create a challenging landscape for maintaining cofactor integrity [62]. Industrial bioprocess conditions particularly exacerbate these challenges, with imperfect mixing leading to oxygen and substrate gradients that propagate cell-to-cell variability. This heterogeneity manifests as plasmid instability, non-expressing subpopulations, and ultimately reduced yield and process stability [62].
Table 1: Environmental Stressors Impacting Cofactor Stability
| Stress Category | Specific Stressors | Impact on Cofactor Function |
|---|---|---|
| Physical | Temperature fluctuations, Imperfect mixing, Shear stress | Protein denaturation, Cofactor dissociation, Altered binding kinetics |
| Chemical | pH shifts, Reactive oxygen species, Metabolic toxins | Oxidative damage, Structural modifications, Cofactor degradation |
| Biological | Proteolytic activity, Metabolic burden, Resource competition | Enzyme degradation, Cofactor depletion, Imbalanced regeneration |
| Process-related | Oxygen limitation, Substrate inhibition, Product accumulation | Reduced cofactor regeneration, Allosteric inhibition, Feedback disruption |
The problem of metabolic burden reflects another fundamental constraint—cellular resources are finite. Excessive heterologous expression sequesters transcription and translation machinery, energy, and precursors including NAD(P)H cofactors [62]. This competition depletes the very cofactor pools essential for engineered enzyme function, creating a self-limiting system where expression of synthetic pathways undermines their own operation. The resulting metabolic burden constrains host growth, diminishes product titers, and accelerates the accumulation of toxic metabolites that further disrupt cofactor stability [62].
Quantitative Analysis of Cofactor Instability
Experimental Measurements of Cofactor-Dependent Activity
Quantifying cofactor instability requires sophisticated analytical approaches that probe both structural and functional integrity. Kinetic assays monitoring hydride transfer provide crucial insights into cofactor functionality. In studies of the aldehyde dehydrogenase Cphy1178, researchers used spectrophotometric measurement of NADH production to characterize activity against short-chain fatty aldehydes [64]. The enzyme displayed optimal activity against propionaldehyde, with a kcat/KM of 1.94 min⁻¹µM⁻¹, but exhibited significant substrate inhibition at high aldehyde concentrations—a phenomenon more pronounced with odd-numbered carbon chains [64]. This substrate inhibition represents a clinically relevant instability mechanism that emerges under specific environmental conditions.
Advanced structural biology techniques have illuminated the molecular basis of cofactor binding instability. X-ray crystallography of Cphy1178 in complex with CoA revealed distinct binding pockets for different cofactor components [64]. The structural analysis showed that the ligand-binding tunnel spans approximately 16Å from the solvent-exposed entry point to the catalytic cysteine, lined with hydrophobic residues that accommodate aldehyde substrates. Mutagenesis studies confirmed that catalytic cysteine (C269) and histidine (H387) residues are essential for activity, with their mutation completely abolishing enzymatic function without destabilizing the overall protein structure [64]. This distinction between catalytic failure and structural collapse highlights the precision required in diagnosing cofactor instability mechanisms.
Table 2: Experimentally Determined Kinetic Parameters of Aldehyde Dehydrogenase Cphy1178
| Substrate | KM (µM) | kcat (min⁻¹) | kcat/KM (min⁻¹µM⁻¹) |
|---|---|---|---|
| Formaldehyde | 1490 ± 310 | 580 ± 40 | 0.39 |
| Acetaldehyde | 300 ± 50 | 870 ± 40 | 2.90 |
| Propionaldehyde | 110 ± 20 | 210 ± 10 | 1.94 |
| Butyraldehyde | 180 ± 30 | 310 ± 20 | 1.70 |
| Valeraldehyde | 410 ± 80 | 340 ± 30 | 0.84 |
Analytical Techniques for Assessing Cofactor Integrity
A multidisciplinary approach is essential for comprehensive cofactor stability assessment. Native mass spectrometry confirmed the tetrameric quaternary structure of Cphy1178, revealing how subunit dissociation could trigger inactivation [64]. Electron spin resonance (ESR) spectroscopy directly detected hydroxyl radicals generated through photocatalytic water splitting, demonstrating how reactive oxygen species contribute to cofactor degradation [65]. Molecular dynamics simulations of hybrid photo-biocatalysts provided additional insights, showing that stable binding complexes between reductive graphene quantum dots (rGQDs) and cross-linked aldo-keto reductase (AKR) form through extensive cation-π and anion-π interactions with interaction strengths of ~14-15 kcal/mol [65].
Chromatographic methods coupled with mass spectrometry enable precise tracking of cofactor integrity during catalytic turnover. For the Cphy1178 enzyme, LC-MS analysis confirmed the presence of propionyl-CoA product, validating the complete catalytic cycle from aldehyde substrate to acyl-CoA product [64]. This approach simultaneously monitors cofactor consumption, product formation, and potential degradation byproducts, offering a comprehensive view of cofactor stability under operational conditions. Zeta potential measurements, XRD, and FT-IR spectroscopy further provide physical characterization of hybrid catalyst systems, revealing aggregation states and chemical bond formation that impact cofactor binding [65].
Stabilization Strategies for Cofactor-Dependent Systems
Protein Engineering and Cofactor Binding Optimization
Strategic engineering of cofactor binding sites represents a powerful approach to enhance stability. Structural analyses inform rational design decisions, as demonstrated by the CoA-bound structure of Cphy1178, which revealed distinct binding pockets for the adenine nucleotides of CoA and NAD+ [64]. This structural insight enables targeted mutations to strengthen cofactor binding without compromising catalytic efficiency. Similarly, molecular dynamics simulations of rGQDs/AKR complexes demonstrated stable binding within 400 ns, with converged RMSD profiles confirming the formation of stable complexes both with and without NADPH cofactor [65]. These computational approaches allow virtual screening of stabilization strategies before experimental validation.
Enzyme engineering efforts have also successfully modified cofactor specificity to enhance stability. In Corynebacterium glutamicum, rational redesign of the coenzyme specificity of glyceraldehyde 3-phosphate dehydrogenase created a de novo NADPH generation pathway that improved lysine production [62]. Such approaches address fundamental cofactor availability constraints by creating self-sufficient cofactor regeneration systems less vulnerable to cellular fluctuations. Similarly, engineering natural and noncanonical nicotinamide cofactor-dependent enzymes expands the toolbox for creating more stable cofactor-enzyme partnerships [62].
Biomolecular Condensates and Microcompartmentalization
Spatial organization offers a sophisticated biological strategy for stabilizing cofactor systems. Bacterial microcompartments (BMCs) are protein-walled metabolic compartments that sequester pathways and maintain private NAD+/NADH cofactor pools isolated from the bulk cytosol [64]. These compartments encapsulate aldehyde dehydrogenase enzymes alongside alcohol dehydrogenase activities to maintain cofactor balance, with the alcohol dehydrogenase recycling NADH produced by the aldehyde dehydrogenase [64]. This spatial coordination ensures that two substrate molecules are processed to produce one acyl-CoA molecule, with the second substrate molecule oxidizing NADH back to NAD+ through alcohol production—an elegant solution to cofactor instability through metabolic coupling.
Eukaryotic systems employ biomolecular condensates formed through liquid-liquid phase separation to create cooperative environments that stabilize transcriptional complexes. These regulatory factor clusters display nonlinear behavior when regulatory factor concentration reaches a critical level, creating abrupt transitions to high-concentration states that stabilize molecular interactions [66]. Context transcription factors establish cooperative environments by mediating enhancer communication and facilitating the formation of these condensates [66]. Synthetic biologists can harness these principles to create stabilized enzymatic environments, using context-only transcription factors that amplify the activity of context-initiator transcription factors despite lacking direct DNA accessibility associations themselves [66].
Diagram 1: Strategic approaches for addressing cofactor instability in engineered enzyme systems.
Hybrid Materials and Cofactor-Independent Systems
The development of cofactor-independent systems represents a paradigm shift in addressing cofactor instability. Recent breakthroughs include hybrid photo-biocatalyst systems based on infrared light and reductive graphene quantum dots (rGQDs) that enable direct hydrogen transfer from water to prochiral substrates without nicotinamide cofactors [65]. These systems mediate synthesis of pharmaceutical intermediates like (R)-1-[3,5-bis(trifluoromethyl)-phenyl] ethanol in 82% yield with >99.99% enantiomeric excess under IR illumination [65]. The rGQDs/AKR photo-biocatalyst assembles through multiple forces (cation-π, anion-π, hydrophobic and π-π interactions) that enable short-range transfer of active hydrogen generated by water splitting under IR illumination to nearby enzyme-bound substrate, completely bypassing traditional cofactor-dependent pathways [65].
The strategic advantage of these systems extends beyond cofactor independence to operational stability. Since the hybrid photo-biocatalysts are insoluble, they can be readily recovered and recycled, addressing both cofactor instability and catalyst reuse challenges [65]. Optical characterization confirms that rGQDs/AKR maintains infrared light responsiveness with upconversion emissions at 525nm, 545nm, and 661nm under 980nm IR light excitation [65]. Bandgap analysis reveals that both rGQDs and rGQDs/AKR exhibit light absorption from UV to infrared regions, with suitable photoredox potentials for water splitting in theory [65]. This innovative approach opens new avenues for creating artificial photo-biocatalyst systems that couple renewable solar energy with sustainable chemical production while circumventing cofactor instability limitations.
Experimental Protocols for Assessing Cofactor Stability
Protocol 1: Kinetic Characterization of Cofactor Dependency
Objective: Quantify enzyme activity and cofactor stability under varying environmental conditions.
Materials:
- Purified enzyme (e.g., aldehyde dehydrogenase Cphy1178)
- Cofactors (NAD+, CoA, etc.)
- Substrates (short-chain aldehydes)
- Assay buffer (typically phosphate or Tris buffer, pH 7.0-8.5)
- Spectrophotometer or plate reader
- LC-MS system for product verification
Method:
- Prepare reaction mixtures containing 50mM Tris-HCl (pH 8.0), 0.2-1.0mM NAD+, 0.1-0.5mM CoA, and varying aldehyde substrates (0.05-2mM) [64].
- Initiate reactions by adding enzyme to final concentration of 0.1-1.0µM.
- Monitor NADH production spectrophotometrically at 340nm for 5-30 minutes.
- Determine kinetic parameters (KM, kcat) by fitting initial velocity data to the Michaelis-Menten equation.
- Verify acyl-CoA product formation by LC-MS: terminate aliquots with equal volume of 0.1% formic acid in acetonitrile, centrifuge, and analyze supernatant using reverse-phase chromatography coupled to mass spectrometry [64].
- Assess cofactor stability by pre-incubating cofactors under different stress conditions (elevated temperature, oxidative stress, pH variations) before activity assays.
Data Analysis: Calculate specific activity, kinetic parameters, and cofactor half-life under stress conditions. Compare intact cofactor concentration before and after stress exposure via HPLC quantification.
Protocol 2: Photo-Biocatalytic Assembly and Testing
Objective: Construct and characterize cofactor-independent photo-enzymatic systems.
Materials:
- Reductive graphene quantum dots (rGQDs)
- Cross-linked enzyme aggregates (CLEAs) of target reductase
- Infrared light source (980nm)
- Reactor with temperature control
- ESR spectroscopy equipment
- Atomic force microscopy
Method:
- Synthesize rGQDs through microwave-assisted method [65].
- Prepare cross-linked aldo-keto reductase (AKR-CLEs) via bio-orthogonal click reaction [65].
- Construct hybrid catalyst by grafting rGQDs onto AKR-CLEs through self-assembly in aqueous solution.
- Characterize assembly using zeta potential measurements, XRD, FT-IR, and AFM to verify successful integration [65].
- Evaluate photocatalytic water splitting: suspend rGQDs/AKR in aqueous substrate solution, illuminate with IR light (980nm) while stirring.
- Monitor hydroxyl radical generation using ESR spectroscopy with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as spin trap [65].
- Quantify reaction products via GC or HPLC: for (R)-3,5-BTPE production, monitor conversion and enantiomeric excess using chiral chromatography [65].
Data Analysis: Calculate product yield, enantiomeric excess, catalyst turnover number, and quantum efficiency. Compare performance to conventional cofactor-dependent system.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Cofactor Stability Research
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Stabilized Enzymes | Cross-linked AKR (AKR-CLEs), Aldehyde dehydrogenase mutants | Engineered for enhanced cofactor binding and resistance to inactivation |
| Advanced Cofactors | Non-canonical nicotinamide analogs, Coenzyme A derivatives | Modified cofactors with improved stability characteristics |
| Nanomaterial Enhancers | Reductive graphene quantum dots (rGQDs), TiO₂ nanotubes | Enable cofactor-independent catalysis or enhanced cofactor regeneration |
| Analytical Standards | Deuterated cofactors, Acyl-CoA analogs, Stable isotope-labeled substrates | Quantification of cofactor stability and metabolic flux analysis |
| Stabilizing Additives | Baicalin (antioxidant), Osmolytes, Polymer encapsulants | Mitigate oxidative stress and stabilize protein structure |
| Molecular Biology Tools | CRISPRi libraries, Expression vectors for cofactor regeneration | Engineer cellular environment for improved cofactor maintenance |
Emerging Solutions and Future Perspectives
The frontier of cofactor stability research points toward increasingly sophisticated integration of materials science with synthetic biology. The successful demonstration of rGQDs as infrared-responsive components in photo-biocatalysts suggests a pathway toward solar-driven biochemical synthesis that completely bypasses traditional cofactor limitations [65]. These systems exploit the abundant conjugate structures with dangling carbon bonds in rGQDs to form stable assemblies with enzymes through multiple weak forces, enabling direct hydrogen transfer from water to substrates [65]. With infrared light responsible for half of solar energy and possessing superior tissue penetration compared to UV/visible light, such systems address both cofactor stability and energy source challenges simultaneously [65].
Future advances will likely focus on orthogonal cofactor systems that operate independently of native metabolic networks, biomimetic compartments that create stabilized microenvironments, and dynamic regulation systems that maintain cofactor homeostasis in response to changing cellular conditions. The discovery that transcription factors can be categorized as "context-only" and "context-initiator" types provides a blueprint for how cooperative environments emerge naturally [66], offering design principles for creating synthetic systems with enhanced stability. As these strategies mature, they will unlock new possibilities in the de novo design of novel enzyme functions, transforming challenges of cofactor instability into engineered features of robust biocatalytic systems.
Optimizing Binding Affinity and Turnover Number via Strategic Mutagenesis
The de novo design of novel enzyme functions represents a frontier in biotechnology, with profound implications for therapeutic development, biocatalysis, and fundamental biological research. A central challenge in this field is the simultaneous optimization of two key enzymatic parameters: binding affinity (often reflected in ( Km )) and catalytic turnover number (( k{cat} )) [67]. These parameters determine the overall catalytic efficiency (( k{cat}/Km )), and optimizing them requires navigating a complex, high-dimensional fitness landscape where mutations can have contrasting effects on stability, substrate binding, and transition state stabilization [68].
Traditional methods, such as directed evolution, have succeeded in improving enzyme functions but often require screening immense libraries of variants, a process that is resource-intensive and time-consuming [67] [68]. The emergence of sophisticated data-driven computational tools is now transforming this paradigm. By enabling the predictive design of enzyme variants, these methods allow researchers to focus experimental efforts on the most promising regions of sequence space, dramatically accelerating the engineering cycle [19] [67] [68]. This technical guide outlines the modern integrative framework—combining deep learning-based protein design, functional prediction, and high-throughput experimental validation—for the strategic optimization of binding affinity and turnover number in the context of de novo enzyme design.
Core Principles of Enzyme Efficiency
The catalytic proficiency of an enzyme is quantized by its efficiency, ( k{cat}/Km ). Optimizing this parameter requires a nuanced understanding of its components:
- Turnover Number (( k_{cat} )): The maximal number of substrate molecules converted to product per enzyme active site per unit time. This rate constant is governed by the activation barrier for the chemical step and is sensitive to mutations that stabilize the transition state.
- Michaelis Constant (( Km )): An approximate measure of the enzyme's binding affinity for its substrate, defined as the substrate concentration at which the reaction rate is half of ( V{max} ). A lower ( K_m ) typically indicates tighter substrate binding.
- Catalytic Efficiency (( k{cat}/Km )): A pseudo-second-order rate constant that describes the enzyme's effectiveness at low substrate concentrations. Improvements often require balancing the sometimes opposing effects of mutations on ( k{cat} ) and ( Km ) [68].
The engineering objective is to traverse the protein fitness landscape via strategic mutagenesis to identify variants where both parameters are favorably altered. This process is complicated by epistasis, where the effect of one mutation depends on the presence of others [68].
Computational Tools for De Novo Enzyme Design
Generative Models for Protein Backbones
RFdiffusion is a generative model based on a fine-tuned RoseTTAFold structure prediction network. It operates as a denoising diffusion probabilistic model to create novel, designable protein backbones from random noise or simple molecular specifications [19].
- Mechanism: The model is trained to iteratively denoise protein backbones corrupted with Gaussian noise, learning to reverse this process to generate realistic structures de novo [19].
- Application in Enzyme Design: RFdiffusion can be conditioned on fixed functional motifs (e.g., active site residues), allowing designers to scaffold a desired catalytic site into a stable, novel protein fold. This is crucial for embedding transition state stabilization networks into a structured environment [19].
Sequence Design and Stability Prediction
Once a stable backbone architecture is generated, the subsequent step is to design a sequence that folds into that structure.
- ProteinMPNN: A neural network that excels at inferring amino acid sequences compatible with a given protein backbone structure. It is typically used downstream of RFdiffusion to generate sequences that are predicted to fold into the designed model [19].
- Stability Prediction with Rosetta: Computational tools like the Rosetta Protein Modeling Suite can calculate the change in folding free energy (( \Delta \Delta G )) upon mutation. This allows for in silico filtering of destabilizing mutations before library construction, dramatically enriching the functional fraction of variants in a library [68].
Table 1: Key Computational Tools for Enzyme Design
| Tool Name | Primary Function | Role in Strategic Mutagenesis |
|---|---|---|
| RFdiffusion [19] | De novo protein backbone generation | Scaffolds functional active sites into novel, stable protein folds. |
| ProteinMPNN [19] | Protein sequence design | Generates sequences that are predicted to fold into a designed backbone structure. |
| Rosetta [68] | Protein energy calculation | Predicts ( \Delta \Delta G ) of mutations to filter out destabilizing variants. |
| HotSpot Wizard [68] | Functional residue identification | Suggests positions for mutagenesis based on sequence and structure analysis. |
Experimental Workflow for Enzyme Optimization
The following diagram illustrates the integrated computational-experimental pipeline for optimizing enzyme kinetics via strategic mutagenesis.
Library Design and Construction
The first experimental stage involves designing and building a high-quality variant library.
- Guided Library Design: Focus mutagenesis on residues within a 6 Å radius of the bound substrate or along substrate access tunnels [68]. Incorporate predictions from tools like HotSpot Wizard and apply a stability filter (e.g., excluding mutations with a predicted ( \Delta \Delta G ) worse than -0.5 Rosetta Energy Units) to avoid non-functional, misfolded variants. This pre-filtering can reduce the experimental library size by approximately 50% without sacrificing beneficial mutations [68].
- Library Construction via Overlap Extension PCR: For comprehensive coverage, synthesize the entire gene using pools of unique DNA oligonucleotides. These oligo pools, typically 200 base pairs in length, are assembled into full-length genes via overlap extension PCR. This method allows for the construction of complex and deep mutant libraries [68].
Screening and Characterization
- High-Throughput Activity Screening: Expression of the variant library in a microbial host like E. coli is followed by screening cell lysates for catalytic activity. For oxidoreductases or hydrolases, this can often be done spectrophotometrically by tracking the appearance of a colored product [68].
- Kinetic Characterization of Hits: Purify the most active hits for detailed biochemical analysis. Determine steady-state kinetic parameters (( k{cat} ) and ( Km )) by measuring initial reaction velocities across a range of substrate concentrations and fitting the data to the Michaelis-Menten model. Software such as KinTek Explorer or ENZO can be used for robust numerical fitting of the time-course data to obtain these parameters [69] [68] [70].
- Structural Validation: Determine high-resolution crystal structures (e.g., 1.5 Å) of optimized variants in complex with substrates or transition state analogs. This provides atomic-level insight into the structural consequences of beneficial mutations, such as improved active site packing, reconfigured hydrogen-bonding networks, or the incorporation of ordered water molecules that stabilize the transition state [68].
Case Study: Evolution of a Kemp Eliminase
The power of this integrated approach is exemplified by the rapid optimization of a de novo-designed Kemp eliminase, HG3 [68]. The experimental results from this study are summarized in the table below.
Table 2: Kinetic Parameters from Kemp Eliminase Optimization (Adapted from [68])
| Enzyme Variant | ( k_{cat} ) (s⁻¹) | ( K_m ) (mM) | ( k{cat}/Km ) (M⁻¹s⁻¹) | Number of Mutations |
|---|---|---|---|---|
| HG3 (Parent) | Not Reported | Not Reported | ~8.0 × 10² | 0 |
| HG3.17 | Not Reported | Not Reported | ~1.7 × 10⁵ | 17 |
| HG3.R5 | 702 ± 79 | ~4.1 × 10⁻³ | 1.7 × 10⁵ | 16 |
Protocol:
- Library Design: All possible single-site mutations in the parent HG3 sequence were analyzed for stability using Rosetta. A library was designed that saturated residues near the active site and tunnel, but included only the ~30% of mutations predicted to be non-destabilizing (( \Delta \Delta G < -0.5 ) REU) [68].
- Gene Synthesis & Screening: The library was constructed via overlap extension PCR from an oligo pool and expressed in E. coli. Activity was screened in cell lysates by monitoring the formation of a colored product at 380 nm [68].
- Iterative Rounds: Beneficial mutations from one round were combined into a combinatorial library to identify the best parent for the subsequent round. This process was repeated for five rounds, culminating in the variant HG3.R5 [68].
- Kinetic Analysis: The best variants from each round were purified, and their steady-state parameters were determined by numerically fitting the total time-course data for substrate depletion [68].
- Structural Analysis: An X-ray structure of HG3.R5 bound to a transition state analog was solved (PDB: 8RD5), revealing the structural basis for improved catalysis, including an ordered water molecule positioned to stabilize the transition state [68].
Outcome: The computationally-guided evolution generated a highly efficient enzyme, HG3.R5, with a >200-fold improvement in catalytic efficiency over the original design. Strikingly, this was achieved in only five rounds of evolution by strategically avoiding destabilizing mutations, thereby enriching the library for functional variants [68].
Table 3: Key Research Reagent Solutions for Enzyme Optimization
| Reagent / Resource | Function and Application |
|---|---|
| Oligo Pools for Gene Synthesis | Enables simultaneous construction of thousands of enzyme variant genes for library generation [68]. |
| Rosetta Software Suite | Provides physics-based and knowledge-based energy functions for predicting protein stability and protein-ligand interactions (( \Delta \Delta G )) [68]. |
| KinTek Explorer / ENZO | Software for simulating complex reaction mechanisms and fitting kinetic data to derive accurate ( k{cat} ) and ( Km ) values [69] [70]. |
| Transition State Analogs (TSAs) | Stable molecules that mimic the transition state of a reaction; used for co-crystallization to visualize and analyze active site geometry [68]. |
The strategic optimization of binding affinity and turnover number is being revolutionized by a new generation of computational methods. The integration of generative models like RFdiffusion for backbone design, sequence prediction tools like ProteinMPNN, and stability filters for intelligent library design creates a powerful and efficient engineering pipeline. The case of the Kemp eliminase HG3.R5 demonstrates that by leveraging these tools to minimize deleterious mutational effects, researchers can rapidly traverse fitness landscapes and achieve remarkable catalytic improvements. This structured, data-driven approach significantly accelerates the de novo design of novel enzymes, paving the way for advanced applications in drug discovery, synthetic biology, and industrial biocatalysis.
Improving Biocompatibility and Shielding from Cellular Metabolites like Glutathione
The de novo design of novel enzyme functions represents a frontier in biotechnology, offering the potential to create custom biocatalysts for therapeutic and diagnostic applications. A central challenge in deploying these designed proteins in vivo is ensuring they remain functional within the complex cellular milieu. The physiological microenvironment is characterized by reductive metabolites, among which glutathione (GSH) is the most abundant intracellular tripeptide antioxidant, typically present at concentrations of 1–10 mM [71] [72]. This "Samsonian life-sustaining small molecule" plays a dual role: it is essential for maintaining cellular redox homeostasis, but its high concentration can impair the function of protein-based therapeutics, particularly those reliant on disulfide bonds or susceptible to thiol-mediated reduction [72]. Furthermore, in specific pathological contexts such as tumors, GSH is overexpressed (∼10 mM, approximately ten times higher than in normal cells), contributing to therapeutic resistance by inactivating reactive oxygen species (ROS)-based treatments and weakening chemotherapeutic agent-induced toxification [73]. Therefore, strategies to improve biocompatibility and shield functional proteins from GSH are critical for advancing the field of de novo enzyme design into practical biomedical applications. This guide synthesizes current knowledge and methodologies, framing them within the overarching research goal of creating robust, designer enzymes for drug development.
Quantitative Data on Glutathione and Shielding Strategies
Understanding the GSH challenge and the performance of various shielding strategies requires a quantitative analysis. The following tables summarize key physiological concentrations and the efficacy of selected intervention approaches.
Table 1: Glutathione Concentrations in Biological Compartments
| Compartment | GSH Concentration | Significance for De Novo Enzymes |
|---|---|---|
| Intracellular Cytosol | 1 - 10 mM [72] | Primary environment for intracellularly delivered enzymes; high reductive pressure. |
| Mitochondria | 10 - 15% of cellular GSH [72] | Critical for enzymes targeting metabolic pathways. |
| Extracellular/Plasma | 10 - 30 μM [72] | Lower threat level for circulating or injectable therapeutics. |
| Cancer Cells | ~10 mM (up to 10x normal) [73] | Creates a highly aggressive reductive environment that necessitates robust shielding. |
Table 2: Efficacy of Selected GSH-Shielding and Depletion Strategies
| Strategy | System/Model | Key Outcome | Reference |
|---|---|---|---|
| N-methylation of GSH Analogues | In vivo pharmacokinetics (rat) | 16.1-fold increase in oral bioavailability; 16.8-fold increase in plasma half-life (t½) [74]. | |
| Metal Nanomaterial GSH Depletion (Cu²⁺) | In vitro (tumor cells) | Direct redox reaction with GSH; activation of Fenton-like reaction with generated Cu⁺ to amplify oxidative stress [73]. | |
| Single-Atom Pd Nanozyme | In vitro (catalytic rate) | Exhibited Michaelis-Menten kinetics in GSH peroxidase-like activity, catalysing GSH oxidation to GSSG [73]. | |
| MgFe₂O₄ NP GSH Depletion (Fe³⁺) | In vitro & In vivo (bone metastasis model) | Fe³⁺ ions react with intracellular GSH to generate Fe²⁺, depleting GSH and enhancing chemodynamic therapy [75]. |
The Glutathione Challenge in Biocompatibility
For a de novo designed enzyme, successful integration into a biological system is dictated by its biocompatibility—the harmonious interaction with the host's physiological environment without eliciting adverse effects [76]. This concept extends beyond mere inertness; for functional enzymes, it encompasses the stability and activity of the protein itself. The "bioactivity zone," the interfacial region between the material surface and the host tissue, is where these interactions are determined [76].
Glutathione directly threatens biocompatibility by disrupting protein structure and function via several mechanisms:
- Reduction of Disulfide Bonds: Many structurally important or catalytically essential disulfide bonds in de novo enzymes are susceptible to reduction by GSH, leading to protein unfolding, aggregation, and loss of function [71] [72].
- Thiol-Mediated Exchange: GSH can participate in thiol-disulfide exchange reactions, potentially leading to the irreversible inactivation of catalytic sites or the formation of incorrect protein adducts [73].
- Attenuation of ROS-Based Mechanisms: For enzymes designed to generate reactive oxygen species (e.g., for catalytic cancer therapies), the high GSH concentration in tumor cells acts as a primary defense mechanism, rapidly scavenging ROS and rendering the treatment ineffective [73] [75].
Overcoming these challenges requires a two-pronged approach: engineering the enzyme itself for intrinsic resilience and employing advanced materials for extrinsic protection.
Strategic Shielding from Glutathione
Intrinsic Engineering of the Protein
Enhancing the innate resistance of a de novo enzyme to GSH involves rational design and modification of its amino acid sequence and structure.
- N-methylation of Peptide Backbones: A highly effective strategy to protect against enzymatic degradation also confers resistance to chemical degradation. The addition of a methyl group to the nitrogen of the peptide bond reduces the molecule's hydrophilicity (fewer hydrogen bond acceptors) and sterically hinders access to the peptide bond, making it less susceptible to GSH-mediated thiol exchange [74]. As evidenced in [74], this modification can dramatically improve pharmacokinetics.
- Alteration of Stereochemistry: Substituting L-amino acids with their D-counterparts in surface-exposed residues can make the protein "invisible" to many native enzymes and less recognizable by GSH and other metabolites, thereby increasing its half-life in vivo [74]. This strategy must be applied judiciously to retain the enzyme's catalytic activity.
- Strategic Placement of Non-Canonical Amino Acids: Incorporating non-natural amino acids that are not substrates for GSH-mediated reactions can shield critical functional regions.
Extrinsic Shielding with Functional Nanomaterials
Nanomaterials can act as protective carriers or shells that isolate the de novo enzyme from the GSH-rich environment until the therapeutic site is reached.
- GSH-Responsive Inorganic Nanomaterials: Materials like manganese dioxide (MnO₂) and copper silicate can be engineered as nanocarriers that degrade specifically in response to high GSH concentrations. This provides programmed release of the encapsulated enzyme within the target cell (e.g., a tumor cell) while offering protection during transit [73]. For instance, MnO₂ nanoshells react with GSH to produce Mn²⁺ and GSSG, effectively depleting the local GSH and triggering the release of the payload [73].
- GSH-Depleting Metal-Organic Frameworks (MOFs) and Nanozymes: An alternative to passive shielding is active GSH depletion. MOFs and single-atom nanozymes (e.g., based on Pd or Cu) can be co-delivered with the therapeutic enzyme. These materials catalytically convert GSH to its oxidized form (GSSG) or consume it in redox reactions, thereby lowering the local GSH concentration and protecting the active enzyme [73] [75]. For example, MgFe₂O₄ nanoparticles release Fe³⁺ ions in the acidic tumor microenvironment, which directly oxidize GSH and also catalyze the production of hydroxyl radicals for combination therapy [75].
The following diagram illustrates the logical decision-making process for selecting an appropriate shielding strategy based on the intended application and cellular environment.
Strategy selection for GSH shielding.
Integrating Shielding into De Novo Enzyme Design Workflows
The principles of GSH shielding must be integrated early into the de novo enzyme design pipeline. The advent of powerful deep-learning-based protein design tools, such as RFdiffusion and ProteinMPNN, allows for the generation of protein structures and sequences from scratch or around specified functional motifs [19]. The shielding strategy can be incorporated as a design constraint.
Diagram: Integrating Shielding into the De Novo Design Workflow
De novo design workflow with GSH shielding.
This workflow highlights the iterative nature of design. The selection of a shielding strategy (Step 5) is informed by the initial design specifications and the in silico performance of the generated enzyme. For instance, if a designed enzyme has a surface-exposed active site, extrinsic shielding with a GSH-responsive nanomaterial might be preferred. Conversely, if the enzyme is intended for intracellular cytosolic action without a carrier, intrinsic engineering via N-methylation might be integrated directly into the sequence design step (Step 3).
Experimental Protocols for Validation
Rigorous experimental validation is essential to confirm the efficacy of any shielding strategy. Below are detailed protocols for key assays.
Protocol: Assessing Enzymatic Stability in a GSH-Rich Environment
Objective: To determine the half-life and residual activity of a de novo designed enzyme after exposure to physiologically relevant concentrations of GSH.
- Reagent Preparation: Prepare a 10 mM GSH solution in a physiologically relevant buffer (e.g., 50 mM Tris-HCl, 1 mM EDTA, pH 7.4). Pre-incubate this solution at 37°C for 15 minutes to allow for thiol equilibration.
- Enzyme Incubation: Mix the de novo enzyme (at a working concentration) with the GSH solution at a 1:1 volume ratio. Include a control sample where the enzyme is mixed with GSH-free buffer.
- Time-Course Sampling: Incubate the mixture at 37°C. Withdraw aliquots at predetermined time points (e.g., 0, 15, 30, 60, 120 minutes).
- Activity Assay: Immediately assay the enzymatic activity of each aliquot using a validated, enzyme-specific activity assay (e.g., spectrophotometric, fluorometric).
- Data Analysis: Plot the residual activity (%) versus time. Calculate the half-life (t½) of the enzyme in the presence and absence of GSH. A successful shielding strategy will show a significantly extended t½ in the GSH group compared to an unshielded control.
Protocol: Quantifying GSH Depletion by Nanozyme Composites
Objective: To measure the rate and extent of GSH depletion by a nanozyme intended for co-delivery with a de novo enzyme.
- Standard Curve: Generate a standard curve for GSH (e.g., 0-1000 μM) using Ellman's reagent (DTNB) according to established protocols.
- Reaction Setup: In a cuvette, combine a known concentration of GSH (e.g., 1 mM, simulating intracellular levels) with the nanozyme (at various concentrations) in reaction buffer. If investigating a peroxidase-mimic, add a low, steady-state concentration of H₂O₂ (e.g., 50-100 μM) [73].
- Kinetic Measurement: Immediately monitor the absorbance of the reaction mixture at 412 nm (for DTNB) over time (e.g., 10-20 minutes) using a spectrophotometer.
- Calculation: Use the standard curve to convert the change in absorbance to the concentration of GSH remaining. Plot [GSH] vs. time to determine the depletion kinetics. Calculate the Michaelis-Menten parameters (Km and Vmax) if the kinetics are saturable, as demonstrated with Pd single-atom nanozymes [73].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for GSH Shielding and Depletion Research
| Reagent / Material | Function / Application | Example Use Case |
|---|---|---|
| Fmoc-Protected D-Amino Acids | Peptide synthesis with altered stereochemistry for intrinsic stability [74]. | Synthesizing GSH-resistant analogues of peptide-based enzymes. |
| Fmoc-Sarcosine (N-methyl glycine) | Introducing N-methylation sites during solid-phase peptide synthesis [74]. | Backbone engineering to block GSH-mediated peptide bond cleavage. |
| MgFe₂O₄ Nanoparticles | Microwave-responsive nanocarrier and GSH-depleting agent (releases Fe³⁺) [75]. | Active GSH depletion in tumor microenvironments for combination therapy. |
| Single-Atom Nanozyme (e.g., Pd) | High-efficiency GSH peroxidase mimic for catalytic GSH oxidation [73]. | Scavenging GSH to protect a co-delivered, GSH-sensitive therapeutic enzyme. |
| GSH Assay Kit (e.g., DTNB-based) | Colorimetric quantification of total and reduced GSH levels [75]. | Measuring GSH depletion efficacy of materials in cell lysates or in vitro. |
| Hollow Mesoporous Silica | Nanocarrier template for constructing GSH-responsive drug delivery systems. | Loading and protecting de novo enzymes for targeted release. |
| Glutathione S-Transferase (GST) | Enzyme for studying GSH conjugation and metabolic pathways. | Validating the resistance of shielded enzymes to GSH-mediated conjugation. |
The journey of a de novo designed enzyme from a computational model to a functional therapeutic agent is fraught with challenges, with the intracellular reductive environment posing a significant barrier. As outlined in this guide, the strategic shielding from glutathione is not an ancillary consideration but a core component of improving biocompatibility and ensuring in vivo efficacy. The integration of intrinsic protein engineering—powered by deep learning tools like RFdiffusion—with advanced extrinsic material science provides a powerful dual-pronged defense. By systematically employing quantitative validation assays and selecting appropriate research tools, scientists can engineer biocatalysts that not only perform novel functions but also survive and thrive within their target biological environment. This synergy between de novo design and metabolic shielding will undoubtedly accelerate the development of next-generation enzyme-based therapeutics.
Balancing Computational Predictions with Experimental Validation and Chemical Intuition
The de novo design of novel enzyme functions represents a frontier in synthetic biology and biotechnology, offering the potential to create custom biocatalysts for applications ranging from therapeutic drug development to green chemistry. This field aims to transcend the limitations of natural evolution by designing proteins with folds and functions not observed in nature [50]. Success in this endeavor hinges on a balanced integration of three core pillars: computational predictions, which leverage artificial intelligence (AI) to explore the vast sequence-space; experimental validation, which grounds designs in physicochemical reality; and chemical intuition, which provides the mechanistic understanding necessary to steer the design process effectively [50] [45]. While AI-driven methods have dramatically accelerated the ability to propose novel protein structures, these designs must navigate the complex energy landscape of protein folding and function. The ultimate test lies in experimental verification, where theoretical designs are synthesized and characterized, often revealing insights that feed back into improved computational models [50]. This guide details the methodologies and frameworks for harmonizing these elements, providing a structured approach for researchers and drug development professionals engaged in creating novel enzymes.
Computational Prediction Methodologies
The computational phase initiates the design process, using AI and physical models to propose viable enzyme candidates from first principles.
AI-Driven Protein Design Frameworks
Modern AI-based methodologies have moved beyond traditional physics-based models like Rosetta, which rely on force-field energy minimization and are often computationally expensive and limited by the accuracy of their energy functions [50]. Instead, machine learning (ML) models trained on vast biological datasets can establish high-dimensional mappings between sequence, structure, and function, enabling the rapid generation of novel, stable proteins [50].
- Generative Models: These models learn the underlying distribution of natural protein sequences and structures, allowing them to propose novel sequences that adhere to biologically plausible patterns. They can be conditioned on specific functional or structural constraints.
- Structure Prediction Tools: The advent of tools like AlphaFold has provided powerful means of validating in silico whether a designed sequence will adopt the intended three-dimensional fold [50].
- Ensemble Methods: Frameworks like SOLVE (Soft-Voting Optimized Learning for Versatile Enzymes) integrate multiple ML models (e.g., Random Forest, LightGBM, Decision Tree) with an optimized weighted strategy. This enhances prediction accuracy for distinguishing enzymes from non-enzymes and predicting Enzyme Commission (EC) numbers [77].
A key advantage of modern ML approaches is their ability to explore regions of the protein functional universe that are inaccessible to natural evolution or traditional directed evolution methods, which are tethered to existing protein scaffolds and perform only local searches in sequence-space [50].
Leveraging Mechanistic Rules for Functional Design
Purely data-driven AI models can sometimes produce designs that are statistically plausible but mechanistically unsound. Incorporating fundamental physicochemical principles is crucial for ensuring proposed enzymes can actually function. Recent research has distilled mechanistic "golden rules" for the de novo design of enzymes, particularly those driven by mechanochemical coupling [45]:
- Friction Matching: The enzyme and its substrate molecule should be attached at the smaller end of each to optimize energy transfer [45].
- Conformational Change Scale: The conformational change of the enzyme must be comparable to or larger than the conformational change required of the substrate molecule [45].
- Rate of Conformational Change: The enzyme's conformational change must be fast enough so that the substrate molecule actually stretches, rather than just following the enzyme without undergoing the required deformation [45].
These rules can be used to inform the training of ML algorithms, fine-tune force fields in all-atom simulations, and provide a critical lens for evaluating computational outputs before proceeding to experimental validation [45].
Table 1: Key Computational Tools and Their Applications in De Novo Enzyme Design
| Tool/Method | Type | Primary Function | Key Advantage |
|---|---|---|---|
| Generative AI Models | Machine Learning | Proposes novel protein sequences and structures | Explores vast, untapped regions of sequence-space beyond natural templates [50]. |
| SOLVE Framework | Interpretable ML | Classifies enzymes and predicts EC numbers from sequence | Uses ensemble learning and provides interpretability via Shapley analysis for functional motifs [77]. |
| Physics-Based (e.g., Rosetta) | Energetic Modeling | Folds proteins and designs active sites via energy minimization | Grounded in physicochemical principles; versatile for rational design [50]. |
| Mechanistic Rules | Theoretical Framework | Guides design for optimal mechanochemical coupling | Provides fundamental physical constraints for functional enzyme design [45]. |
Diagram 1: The iterative computational prediction workflow for generating candidate enzyme sequences.
Experimental Validation Protocols
Computational designs must be rigorously tested in the laboratory. This phase confirms the protein's structure, stability, and catalytic function.
Structural and Biophysical Characterization
The first step is to verify that the synthesized protein folds into the intended three-dimensional structure.
- Gene Synthesis and Cloning: The designed nucleotide sequence is synthesized and cloned into an appropriate expression vector.
- Protein Expression and Purification: The protein is expressed in a host system (e.g., E. coli) and purified using chromatography techniques (e.g., affinity, size-exclusion).
- Circular Dichroism (CD) Spectroscopy: This technique assesses the secondary structure composition (e.g., alpha-helical, beta-sheet content) and thermal stability of the designed protein.
- X-ray Crystallography/Cryo-Electron Microscopy (Cryo-EM): These high-resolution methods provide atomic-level detail of the protein's structure, allowing for direct comparison with the computational model.
- Nuclear Magnetic Resonance (NMR) Spectroscopy: NMR can provide information on both structure and dynamics in solution.
Functional Activity Assays
Confirming the presence of the intended structure is necessary but not sufficient; the enzyme must also perform its designed catalytic function.
- Steady-State Kinetic Assays: These assays measure the catalytic efficiency of the enzyme by determining parameters such as the turnover number (k~cat~) and the Michaelis constant (K~m~). This involves monitoring the formation of product or consumption of substrate over time under saturating conditions.
- Surface Plasmon Resonance (SPR) or Isothermal Titration Calorimetry (ITC): These techniques quantify binding affinity (K~d~) between the enzyme and its substrate or inhibitor, which is critical for validating designed molecular recognition interfaces.
- In vivo Functional Complementation: For enzymes designed to function in a biological context, complementation assays in knockout strains can validate function within a cellular environment.
Table 2: Core Experimental Techniques for Validating De Novo Enzymes
| Technique | Property Measured | Typical Experimental Protocol |
|---|---|---|
| Circular Dichroism (CD) | Secondary structure, thermal stability | Measure far-UV spectrum (190-250 nm);\nPerform thermal denaturation while monitoring ellipticity at 222 nm. |
| Size-Exclusion Chromatography with Multi-Angle Light Scattering (SEC-MALS) | Oligomeric state, solution molecular weight | Inject purified protein onto SEC column inline with MALS and refractive index detectors. |
| Steady-State Kinetics | Catalytic efficiency (k~cat~/K~m~) | Incubate enzyme with varying substrate concentrations;\nMonitor product formation spectrophotometrically or chromatographically. |
| X-ray Crystallography | Atomic-resolution 3D structure | Grow protein crystals, collect diffraction data, solve and refine structure. |
Integrating Chemical Intuition and Iterative Learning
The most powerful design cycles are those where computational design and experimental testing are tightly coupled, with chemical intuition guiding the interpretation of results to inform the next design iteration.
The Role of Interpretable AI
The "black box" nature of some complex ML models can be a barrier to understanding why a design succeeds or fails. Using interpretable ML approaches is critical for building chemical intuition. For instance, the SOLVE framework employs Shapley analysis to identify which specific subsequences or motifs in the primary sequence are most influential for the predicted function [77]. This allows researchers to move beyond mere prediction to gain insights into the structural and mechanistic basis of enzyme activity, which can then be applied to refine future design rules.
Analyzing and Learning from Design Failures
Not all computationally designed enzymes will fold or function as intended. Misfolded proteins, aggregated species, and inactive designs are common initial outcomes. A systematic analysis of failures is a rich source of learning.
- Characterize Misfolded States: Use techniques like SEC and analytical ultracentrifugation to identify aggregation, and NMR or CD to probe for disordered structures.
- Analyze Active Site Geometry: For proteins that fold but lack activity, compare the crystal structure of the designed enzyme with the computational model. Inaccuracies in side-chain packing, hydrogen bonding networks, or electrostatic environments in the active site are often the culprit.
- Feed Data Back into Models: The experimental data from failed designs is invaluable for retraining and improving ML models. For example, negative design data (sequences that do not fold or function) can help the model learn the boundaries of the viable sequence-structure space more accurately.
Diagram 2: The iterative design cycle, where experimental results and human intuition refine computational models.
The Scientist's Toolkit: Research Reagent Solutions
A successful de novo enzyme design pipeline relies on a suite of specialized reagents, software, and databases. The following table details key resources essential for the featured experiments and computational work.
Table 3: Essential Research Reagents and Tools for De Novo Enzyme Design
| Item | Function/Description | Application in Workflow |
|---|---|---|
| Gene Synthesis Services | Provides custom double-stranded DNA fragments encoding the designed protein sequence. | Critical first step for moving from in silico sequences to physical proteins for expression and testing. |
| Heterologous Expression Systems (e.g., E. coli, insect cells) | Living cells used as factories to produce large quantities of the designed protein. | Protein production for biophysical characterization and activity assays. |
| Affinity Chromatography Resins (e.g., Ni-NTA) | Matrices for purifying proteins based on specific tags (like polyhistidine) fused to the designed protein. | Rapid, high-purity isolation of the expressed enzyme from cell lysates. |
| Spectrophotometer / Plate Reader | Instrument for measuring light absorption or emission of samples in cuvettes or microplates. | Essential for running kinetic assays to monitor enzyme activity by tracking substrate depletion or product formation. |
| AlphaFold Protein Structure Database | Repository of predicted protein structures for millions of sequences [50]. | Provides a reference for comparing designed novel folds and for sourcing data for training computational models. |
| SOLVE Software | An interpretable machine learning tool for enzyme function prediction from primary sequence [77]. | Used to predict the potential function of newly designed sequences and identify key functional motifs. |
Benchmarking Success: Validation Frameworks and Comparative Analysis of De Novo Enzymes
In the field of de novo enzyme design, the ultimate proof of a successful design lies in its measurable function. The creation of novel proteins with prescribed activities from scratch represents a frontier in biological engineering, enabled by powerful new deep-learning methods like RFdiffusion [19]. However, a designed enzyme's three-dimensional structure is merely a starting point; its true utility for research, therapeutics, or industrial processes depends on quantitative performance metrics. Turnover number (TON), binding affinity (KD), and catalytic efficiency form a fundamental triad of parameters that provide a rigorous assessment of an enzyme's functional capability.
This guide details these core metrics within the modern context of de novo enzyme design. The advent of artificial intelligence (AI) trained on vast datasets of protein sequences and structures has revolutionized the field, allowing researchers to "write" proteins with new shapes and functions without starting from natural templates [78]. As we progress toward designing enzymes for bespoke chemical transformations, molecular recognition, and synthetic cellular signaling, a deep understanding of these quantitative metrics is paramount for validating and iterating on computational designs.
Defining the Core Metrics
Turnover Number (TON) and Turnover Frequency (TOF)
The term "turnover number" has two distinct yet related meanings in chemistry and enzymology, which must be clearly differentiated.
In Enzymology (kcat): The turnover number (kcat) is defined as the limiting number of chemical conversions of substrate molecules per second that a single active site can execute when the enzyme is saturated with substrate [79] [80]. It is calculated from the maximum reaction rate ((V{max})) and the total concentration of active sites ((e0)): [ k{\mathrm{cat}} = \frac{V{\max}}{e_0} ] For enzymes with a single active site, kcat is also referred to as the catalytic constant [79]. This value represents the intrinsic speed of the catalytic cycle once substrate binding is no longer rate-limiting.
In Organometallic and Industrial Catalysis (TON): The turnover number (TON) refers to the total number of moles of substrate that a mole of catalyst can convert before it becomes inactivated [79]. It is a measure of a catalyst's lifetime and total productivity: [ \mathrm{TON} = \frac{n{\mathrm{product}}}{n{\mathrm{cat}}} ] An ideal catalyst would have an infinite TON, as it would never degrade.
Turnover Frequency (TOF): This metric connects the two concepts, defined as the turnover per unit time (e.g., TON per second). It is equivalent to the meaning of turnover number in enzymology and provides a rate of catalytic activity [79]. For most industrial applications, TOF values typically range from 10−2 to 102 s−1, while enzymes can achieve dramatically higher frequencies, up to 107 s−1 for catalase [79].
Table 1: Key Metrics for Catalytic Activity
| Metric | Symbol | Definition | Typical Range | Primary Significance |
|---|---|---|---|---|
| Turnover Number (Enzymology) | (k_{cat}) | Substrate conversions per active site per second at saturation [79]. | Up to 4×10⁷ s⁻¹ (catalase) [79] | Intrinsic speed of the catalytic step. |
| Turnover Number (Catalysis) | TON | Total moles of substrate converted per mole of catalyst over its lifetime [79]. | Varies widely; ideal is infinite. | Total productivity and lifetime of catalyst. |
| Turnover Frequency | TOF | TON per unit time [79]. | 10⁻² – 10² s⁻¹ (industrial), up to 10⁷ s⁻¹ (enzymes) [79] | Rate of catalytic activity. |
Binding Affinity (KD)
The dissociation constant (KD) is an equilibrium constant that quantifies the binding affinity between two molecules, such as an enzyme and its substrate or a drug and its receptor. It is defined as the concentration of ligand at which half of the binding sites on a partner are occupied at equilibrium [81] [82].
A lower KD value indicates tighter binding (higher affinity), as less ligand is required to achieve half-saturation. Conversely, a higher KD indicates weaker binding. KD values can span a vast range, from picomolar (pM, 10⁻¹² M) for very tight interactions like high-affinity antibody-antigen pairs, to micromolar (μM, 10⁻⁶ M) or even higher for weaker interactions, such as some enzyme-substrate complexes [81] [82].
Conceptually, for a simple bimolecular interaction: [ R + L \rightleftharpoons RL \quad \text{and} \quad K_D = \frac{[R][L]}{[RL]} ] Where (R) is the receptor (e.g., enzyme), (L) is the ligand (e.g., substrate), and (RL) is the complex.
Catalytic Efficiency (kcat/KM)
While kcat describes the catalytic rate at saturation, and KD describes binding affinity, the catalytic efficiency, defined by the ratio (k{cat}/KM), is a more holistic metric. It combines both binding and catalytic steps into a single parameter [83] [84].
The Michaelis constant ((KM)) is the substrate concentration at which the reaction rate is half of (V{max}). While not identical to KD, it is often related and provides a measure of the apparent substrate affinity. The ratio (k{cat}/KM) is a second-order rate constant that describes the enzyme's efficiency at low substrate concentrations [83].
The upper limit for (k{cat}/KM) is set by the rate of diffusion, typically between 10⁸ to 10⁹ M⁻¹s⁻¹ [80]. Enzymes like triose phosphate isomerase, which have ratios approaching this range, are considered to have achieved "catalytic perfection" [80]. This metric is also known as the specificity constant, as it can be used to compare an enzyme's preference for different substrates [80].
Table 2: Summary of Core Enzyme Performance Metrics
| Metric | Formula | Interpretation | Theoretical Limit |
|---|---|---|---|
| Catalytic Efficiency | (k{cat}/KM) | Efficiency at low substrate concentrations; specificity for a substrate [80]. | ~10⁸ – 10⁹ M⁻¹s⁻¹ (diffusion limit) [80]. |
| Michaelis Constant | (K_M) | Substrate concentration at half-maximal velocity; an inverse measure of apparent affinity. | Not applicable. |
| Dissociation Constant | (K_D) | Equilibrium constant for complex dissociation; a direct measure of binding affinity [81]. | Not applicable. |
Experimental Protocols for Measurement
Determining Turnover Number (kcat)
The protocol for determining the enzymatic turnover number (kcat) relies on measuring the enzyme's activity under saturating substrate conditions.
Principle: kcat is derived from the maximum reaction velocity ((V{max})) achieved when the enzyme is fully saturated with substrate, according to the equation (k{cat} = V{max} / e0), where (e_0) is the molar concentration of active enzyme sites [79].
Procedure:
- Prepare Substrate Titration: Create a series of reactions with a fixed, known concentration of enzyme and varying concentrations of substrate, ensuring the highest concentrations are significantly above the expected (K_M) to achieve saturation.
- Measure Initial Velocity: For each substrate concentration, measure the initial velocity ((v_0)) of the reaction by tracking the appearance of product or disappearance of substrate over time. This can be done via spectrophotometry, fluorometry, or other suitable techniques.
- Plot Michaelis-Menten Curve: Plot (v0) against substrate concentration ([S]). The curve will hyperbolic, plateauing at (V{max}).
- Nonlinear Regression Analysis: Fit the data to the Michaelis-Menten equation, ( v0 = \frac{V{max}[S]}{KM + [S]} ), to determine (V{max}) accurately.
- Calculate kcat: Divide the obtained (V{max}) by the total concentration of active enzyme ((e0)) to calculate kcat [79] [83].
Measuring Binding Affinity (KD)
A wide range of technologies can be used to determine the dissociation constant (KD). The general principle involves incubating a fixed concentration of one binding partner with a titration of the other and measuring a concentration-dependent change in a signal.
Protocol using Microfluidic Diffusional Sizing (MDS) [81]:
- Labeling: A binding partner (the "probe") is fluorescently labelled.
- Titration: The labelled probe is incubated with increasing concentrations of the unlabeled ligand.
- Measurement: The mixture is subjected to MDS, which measures the change in the hydrodynamic radius ((R_h)) of the probe that occurs upon binding and complex formation.
- Data Analysis: The change in size ((R_h)) is plotted against the ligand concentration. The KD is determined as the ligand concentration at the inflection point of this binding curve, where half of the probe is bound [81].
Important Considerations:
- Buffer Conditions: KD is highly dependent on experimental conditions like pH, ionic strength, and temperature. These must be carefully controlled and reported [81].
- Binding Model: A simple 1:1 binding model is often assumed, but many biological interactions are more complex (e.g., cooperativity, multiple sites). Using an incorrect model will lead to an inaccurate KD [81].
Assessing Catalytic Efficiency (kcat/KM)
The catalytic efficiency is not measured directly but is derived from the same experimental data used to determine kcat.
- Procedure:
- Perform Michaelis-Menten Experiment: As described in Section 3.1, conduct a substrate titration experiment and fit the data to the Michaelis-Menten equation to obtain both (V{max}) and (KM).
- Calculate Active Enzyme Concentration: Determine the molar concentration of active enzyme sites ((e0)) in the assay. This is a critical and often challenging step, as impurities or inactive protein can lead to underestimation of kcat.
- Compute Metrics: Calculate (k{cat} = V{max} / e0).
- Determine Catalytic Efficiency: Calculate the ratio (k{cat}/KM) [83] [84].
This workflow for determining catalytic efficiency is summarized in the following diagram:
Metrics in De Novo Enzyme Design
The transition from designing stable protein structures to creating functional enzymes is a central challenge in de novo protein design. Quantitative metrics like TON, KD, and kcat/KM are the critical benchmarks for success in this endeavor.
AI-based generative methods, such as RFdiffusion, are revolutionizing the field. This method fine-tunes the RoseTTAFold structure prediction network on protein structure denoising tasks, creating a generative model that can produce novel protein backbones from random noise [19]. This allows for the design of proteins with new shapes and the scaffolding of functional sites, such as enzyme active sites, into these novel structures [19]. The functional quality of these designs is then validated by the metrics discussed in this guide.
A key application is the design of protein binders. In one landmark demonstration, an RFdiffusion-designed binder targeting influenza haemagglutinin was experimentally characterized. Its cryogenic electron microscopy (cryo-EM) structure in complex with the target was nearly identical to the computational design model, a feat that underscores the method's atomic-level accuracy [19]. For such a binder, a low KD value would be a primary quantitative measure of its success.
Furthermore, the integration of AI extends beyond structure generation to the direct optimization of function. Machine learning models are now being trained on large biochemical datasets to predict mutations that enhance catalytic efficiency (kcat/KM), Thermostability, and other desirable properties, significantly accelerating the enzyme engineering cycle [85].
The following diagram illustrates how computational design and experimental validation are integrated in a functional design cycle.
The Scientist's Toolkit: Essential Research Reagents and Methods
Success in de novo enzyme design and characterization relies on a suite of specialized reagents, tools, and methodologies.
Table 3: Key Reagents and Tools for Enzyme Design and Characterization
| Tool / Reagent | Function / Description | Relevance to Metrics |
|---|---|---|
| RFdiffusion [19] | A generative AI model based on RoseTTAFold for de novo design of protein backbones and scaffolding of functional motifs. | Generates novel enzyme designs for functional testing. |
| ProteinMPNN [19] | A neural network for designing amino acid sequences that fold into a given protein backbone structure. | Provides sequences for computationally designed enzymes. |
| Microfluidic Diffusional Sizing (MDS) [81] | An in-solution technique that measures changes in hydrodynamic radius (R_h) upon binding to determine affinity and stoichiometry. | Measures K_D in solution under native conditions. |
| Surface Plasmon Resonance (SPR) [81] | A surface-based technique that measures biomolecular interactions in real-time without labels. | A traditional method for determining K_D and binding kinetics. |
| AlphaFold2 / ESMFold [19] | Protein structure prediction networks; used for in silico validation of designed structures. | Validates that a designed sequence will fold into the intended structure. |
| Directed Evolution Platforms [85] | A pipeline for creating and screening mutant libraries to improve enzyme properties like activity or stability. | Used to optimize initial designs to improve kcat and kcat/K_M. |
The quantitative metrics of turnover number, binding affinity, and catalytic efficiency are indispensable for translating the abstract outputs of de novo protein design into tangible, functional enzymes. As the field progresses, driven by AI tools like RFdiffusion, the role of these metrics becomes ever more critical. They provide the rigorous, quantitative feedback needed to close the design loop, informing subsequent rounds of computational design and engineering.
The future of de novo enzyme design lies in the seamless integration of predictive computational modeling, high-throughput experimental characterization, and iterative optimization based on these fundamental performance parameters. By grounding the assessment of novel designs in these robust quantitative metrics, researchers can continue to push the boundaries of what is possible, creating bespoke enzymes for applications in therapeutics, green chemistry, and synthetic biology.
The de novo design of novel enzyme functions represents a frontier in biotechnology, with applications ranging from sustainable synthesis to therapeutic development. The central challenge in this field lies in transitioning from initial design concepts to highly efficient, stable catalysts without relying on exhaustive experimental screening. Computational benchmarking is crucial for evaluating and refining these designs, providing atomistic insights into catalytic efficiency and stability. Among various simulation methods, the Empirical Valence Bond (EVB) approach has emerged as a powerful tool for quantitative prediction of enzymatic activity. This guide examines the role of EVB and complementary computational methods in benchmarking and advancing de novo enzyme design, providing researchers with protocols and frameworks for their implementation.
Methodological Foundations of Key Simulation Approaches
The Empirical Valence Bond (EVB) Method
The EVB method is a quantum-mechanics/molecular-mechanics (QM/MM) approach that models chemical reactions by representing the system through resonance structures or diabatic states corresponding to classical valence-bond structures. These states are mixed to describe the reacting system [86]. The methodology employs a Hamiltonian matrix where the diagonal elements (H₁₁ and H₂₂) represent classical force fields for the reactant and product states of the reaction, while the off-diagonal element (H₁₂) represents the coupling between these states [87].
A key strength of EVB is its parametrization strategy. The method is calibrated using experimental or quantum chemical data from a reference reaction in solution, typically targeting the activation free energy (ΔG‡) and reaction free energy (ΔG₀). This is achieved by adjusting the coupling element H₁₂ and the energy difference Δα between the diabatic states [87]. Once parametrized, the same EVB potential is transferred to the enzyme environment without further adjustment, allowing for direct prediction of catalytic effects [86]. This robust parametrization enables EVB to accurately reproduce experimental activation enthalpies and entropies, as demonstrated for enzymes like ketosteroid isomerase and chorismate mutase [87].
Complementary Computational Chemistry Methods
Other computational methods play complementary roles in enzyme design and benchmarking:
Quantum Chemical Methods: Density Functional Theory (DFT) and coupled-cluster techniques provide accurate mapping of potential energy surfaces and characterization of transition states [88]. These methods are essential for developing initial "theozyme" designs—theoretical catalytic sites optimized for the transition state [6].
Hybrid QM/MM Models: These approaches combine quantum mechanical treatment of the active site with molecular mechanics description of the protein environment, offering a balanced compromise between accuracy and computational cost for studying enzyme reactions [88].
Machine Learning (ML) and Artificial Intelligence (AI): Recent advances include models like TopEC, a 3D graph neural network that predicts enzyme function from structure by focusing on atomic environments around active sites [89]. ML methods enhance physics-based modeling by performing dimension reduction on complex molecular dynamics datasets and identifying catalytically relevant modes [90].
Table 1: Key Computational Methods for Enzyme Design Benchmarking
| Method | Primary Function | Strengths | Limitations |
|---|---|---|---|
| Empirical Valence Bond (EVB) | Calculate activation free energies and mutational effects | Quantitative prediction of ΔG‡, ΔH‡, ΔS‡; less computationally demanding than other QM/MM methods | Requires careful parametrization against reference reactions |
| Density Functional Theory (DFT) | Electronic structure calculation, transition state characterization | Favorable balance of accuracy and efficiency for medium-sized systems | Reliability depends on functional choice; challenged by strong correlation |
| Hybrid QM/MM | Simulate reactions in protein environments | Quantum detail for active site with larger-scale context | Setup complexity; QM/MM boundary effects |
| Machine Learning (e.g., TopEC) | Function prediction from structure | High-throughput screening; recognizes similar functions across different structures | Dependent on training data quality and quantity |
Benchmarking EVB Performance in Enzyme Design
Quantitative Assessment of Catalytic Efficiency
EVB has demonstrated remarkable accuracy in predicting the effects of mutations on catalytic activity. In a benchmark study on chorismate mutase, EVB calculations reproduced experimental activation free energies within ~2 kcal/mol for multiple variants [86]. The table below shows the close agreement between calculated and observed activation barriers:
Table 2: EVB Performance in Predicting Activation Barriers for Chorismate Mutase Variants [86]
| Enzyme Variant | Calculated Δg‡cat (kcal/mol) | Observed Δg‡cat (kcal/mol) | Difference (kcal/mol) |
|---|---|---|---|
| EcCM (native) | 15.3 ± 1.5 | 15.3 | 0.0 |
| V35I-EcCM | 13.3 ± 0.6 | 15.0 | -1.7 |
| V35A-EcCM | 15.2 ± 0.9 | 15.7 | -0.5 |
| mMjCM (monomer) | 16.2 ± 1.7 | 16.8 | -0.6 |
| BsCM (native) | 16.6 ± 1.6 | 15.3 | +1.3 |
| R90Cit-BsCM | 23.7 ± 2.5 | 21.1 | +2.6 |
| R90G-BsCM | 23.8 ± 2.2 | 22.5 | +1.3 |
Application to Kemp Eliminase Design
The Kemp elimination reaction has served as a benchmark for de novo enzyme design due to its simplicity and absence of natural enzymes catalyzing this reaction [91] [6]. EVB has proven particularly valuable in optimizing de novo Kemp eliminases. In one study, EVB simulations reproduced experimental activation energies for optimized eliminases to within ~2 kcal mol⁻¹, revealing that enhanced activity was linked to better geometric preorganization of the active site [91].
Recent breakthroughs in fully computational enzyme design have produced Kemp eliminases with catalytic efficiencies exceeding 10⁵ M⁻¹ s⁻¹ and turnover numbers of ~30 s⁻¹, rivaling natural enzymes [6]. These designs incorporated novel active sites with over 140 mutations from any natural protein, achieving high stability (>85°C) without requiring mutant-library screening [6]. EVB provided critical validation during the design process, confirming the catalytic competence of the proposed active site arrangements.
Experimental Protocols for EVB Implementation
Standard EVB Simulation Workflow
The following diagram illustrates the comprehensive workflow for implementing EVB simulations in enzyme design projects:
EVB Simulation Workflow for Enzyme Design
Detailed Simulation Protocol
Step 1: System Preparation
- Obtain coordinates for the enzyme-substrate complex from crystallography, homology modeling, or de novo design
- Solvate the system using an appropriate water model (e.g., SCAAS model with 18 Å radius water sphere)
- Apply surface constraints and treat long-range electrostatic effects using methods like the Local Reaction Field (LRF) [86]
Step 2: EVB Region Definition
- Select atoms for inclusion in the EVB region, typically comprising the entire substrate and key catalytic residues (e.g., 24 atoms for chorismate mutase) [86]
- Define the diabatic states (reactant and product) based on quantum chemical optimization of structures in the gas phase using methods like B3LYP/6-31+G(d) [86]
Step 3: Parametrization of the EVB Hamiltonian
- Perform quantum mechanical calculations to derive charges for the reactant and product states
- Calibrate H₁₂ and Δα parameters to reproduce the observed activation barrier for the reference reaction in solution
- Use identical EVB parameters for both solution and enzyme environments without further adjustment [86]
Step 4: Free Energy Calculation via FEP/Umbrella Sampling
- Implement a mapping potential εₘ = (1-θₘ)ε₁ + θₘε₂ where θₘ changes from 0 to 1 in fixed increments [86]
- For each window (typically 21 frames), run MD simulations (20 ps each) with a time step of 1 fs at 300K [86]
- Repeat simulations multiple times (≥5) with different initial conditions to ensure statistical reliability [86]
- Calculate free energy profiles using umbrella sampling and the weighted histogram analysis method (WHAM)
Step 5: Data Analysis and Validation
- Construct computational Arrhenius plots from temperature-dependent ΔG‡ values to extract ΔH‡ and ΔS‡ [87]
- Compare predicted activation barriers with experimental kinetic data (kcat, KM)
- Use insights from EVB simulations to guide redesign, focusing on residues that impact transition state stabilization
Integrated Computational Workflows in Enzyme Design
The integration of EVB with other computational methods has enabled more robust enzyme design pipelines. The following diagram illustrates how EVB fits into a comprehensive computational workflow for de novo enzyme design:
Integrated Computational Enzyme Design Pipeline
This integrated approach combines multiple computational strategies:
- Theozyme Design: Quantum mechanical calculations define optimal transition state geometry and interactions [6]
- Scaffold Selection and Backbone Generation: Natural protein folds (e.g., TIM-barrel) or de novo designed backbones provide structural frameworks [6]
- Sequence Design: Methods like Rosetta and PROSS optimize sequences for stability and foldability [6]
- Active Site Optimization: FuncLib uses phylogenetic analysis and Rosetta design to suggest stable, diverse active site variants [91]
- Catalytic Assessment: EVB provides quantitative prediction of catalytic efficiency before experimental testing [91] [86]
Essential Research Reagents and Computational Tools
Successful implementation of EVB and related methods requires specific computational tools and resources:
Table 3: Essential Research Reagent Solutions for Computational Enzyme Design
| Tool/Resource | Type | Primary Function | Application in Enzyme Design |
|---|---|---|---|
| MOLARIS | Software Package | EVB/MD simulations | Calculate activation free energies and mutational effects [86] |
| Rosetta | Software Suite | Protein design & structure prediction | Scaffold design and active site optimization [91] [6] |
| Gaussian | Quantum Chemistry | Electronic structure calculations | Parametrize EVB diabatic states; theozyme design [86] |
| FuncLib Web Server | Web Resource | Active site redesign | Generate stable, diverse enzyme variants [91] |
| TopEC | Machine Learning Model | Enzyme function prediction | Predict Enzyme Commission classes from 3D structure [89] |
| JUWELS Supercomputer | HPC Infrastructure | Large-scale computation | Train ML models; run high-throughput simulations [89] |
The Empirical Valence Bond method has established itself as a powerful tool for computational benchmarking in de novo enzyme design, providing quantitative predictions of catalytic activity that guide experimental efforts. When integrated with other computational approaches—including quantum chemistry, machine learning, and protein design algorithms—EVB contributes to robust workflows that accelerate the creation of novel biocatalysts. Recent successes in designing highly efficient Kemp eliminases demonstrate the maturity of these integrated computational strategies, offering a paradigm for developing enzymes that catalyze new-to-nature reactions with efficiencies rivaling natural enzymes. As computational power increases and methods refine further, the role of physics-based modeling approaches like EVB will continue to expand, enabling more ambitious enzyme design projects with reduced experimental optimization.
The de novo design of novel enzyme functions represents a frontier in synthetic biology, holding promise for creating bespoke biocatalysts, therapeutics, and solutions for environmental sustainability. This field is currently defined by two dominant computational paradigms: established physics-based modeling tools, exemplified by the Rosetta software suite, and emerging AI-driven generative workflows, such as those powered by RFdiffusion and ProteinMPNN. The former relies on thermodynamic principles and biological knowledge, while the latter leverages patterns learned from vast datasets of protein sequences and structures. This whitepaper provides a comparative analysis of these strategies, examining their underlying principles, methodological workflows, performance, and practical applications within enzyme design. The objective is to equip researchers and drug development professionals with a technical framework for selecting and implementing these powerful technologies in their de novo design projects.
Core Principles and Evolutionary Trajectory
Rosetta: A Physics-Based and Knowledge-Based Modeling Approach
Rosetta is a comprehensive macromolecular modeling software whose development spans over two decades, driven by a global community of laboratories [92]. Its core principle is grounded in Anfinsen's hypothesis, which posits that a protein's native structure corresponds to its global free energy minimum [50]. Rosetta operationalizes this by combining physics-based energy calculations with knowledge-based statistical potentials derived from high-resolution crystal structures [92].
Its energy function, REF2015, is a linear combination of weighted terms representing van der Waals forces, hydrogen bonding, electrostatics, solvation, and backbone torsion preferences [92]. Conformational sampling is typically achieved through stochastic methods like Monte Carlo with simulated annealing, guided by the Metropolis criterion to accept or reject new poses based on their energy [92]. While powerful, this approach has inherent limitations. The force fields are approximations, and minor inaccuracies can lead to designs that misfold experimentally. Furthermore, the computational expense of exhaustively sampling sequence and structure space is prohibitive, particularly for large or complex proteins [50].
AI-Driven Workflows: A Data-Centric Generative Paradigm
AI-driven de novo protein design constitutes a paradigm shift from energy minimization to data-driven generation [50] [78]. These methods use deep learning models—including generative adversarial networks, variational autoencoders, and most recently, diffusion models—trained on millions of protein sequences and structures. They learn high-dimensional mappings between sequence, structure, and function, allowing them to generate novel, stable, and functional proteins that explore regions of the protein universe untouched by natural evolution [50] [93].
A landmark model in this space is RFdiffusion, which adapts the RoseTTAFold structure prediction network into a denoising diffusion probabilistic model [19]. It generates protein backbones by iteratively denoising a cloud of random residue frames, a process that can be conditioned on simple molecular specifications like a binding site or a symmetric architecture [19]. ProteinMPNN is another critical tool, a neural network that efficiently designs sequences for given protein backbones, solving the inverse folding problem with high success rates [19]. This AI-driven approach fundamentally expands the possibilities within protein engineering by freeing it from a reliance on natural templates [78].
Table 1: Foundational Principles of Rosetta and AI-Driven Design Strategies.
| Feature | Rosetta | AI-Driven Workflows |
|---|---|---|
| Core Principle | Thermodynamic stability (global free energy minimum) [50] [92] | Statistical patterns from data; generative modeling [50] [19] |
| Methodological Basis | Physics-based & knowledge-based force fields; Monte Carlo sampling [92] | Deep learning (e.g., diffusion models, protein language models) [19] [94] |
| Training Data | High-resolution crystal structures for statistical potentials [92] | Large-scale sequence (e.g., UniProt) and structure (e.g., PDB) databases [50] |
| Key Strength | High interpretability; precise control over atomic-level interactions [92] | Unprecedented speed and diversity in exploring novel sequence-structure space [50] [19] |
| Inherent Limitation | Computationally expensive sampling; approximate force fields [50] | "Black box" nature; limited interpretability; performance tied to training data [50] |
Comparative Workflow Analysis
The process of de novo enzyme design, from concept to validated construct, involves distinct stages. The following diagram illustrates the typical workflows for both Rosetta and AI-driven approaches, highlighting key divergences.
The Rosetta de novo Design Protocol
The traditional Rosetta protocol is complex and often described as "more art than science," requiring significant expertise and iterative tuning [95]. A typical workflow for a novel enzyme fold involves:
- Backbone Blueprint Creation: The designer manually defines the target protein's architecture using a blueprint file, specifying secondary structure elements (SSEs)—helices (H), strands (E), and loops (L)—and their chirality [95]. For complex topologies, the protein may be split into segments (e.g., two halves) designed separately to manage sampling complexity [95].
- Backbone Construction: Using movers like
BluePrintBDR, Rosetta assembles a backbone conformer. This involves cyclic coordinate descent (CCD) for loop closure and fragment insertion to sample conformations, guided by the score function [92] [95]. - Sequence Design: With a fixed backbone, the sequence is optimized using a Monte Carlo protocol to sample amino acid identities. This is heavily constrained by resfiles that dictate designable positions and allowed amino acids (e.g.,
PIKAAto pick a specific amino acid,NOTAAto exclude certain types) to enforce functional site identities and prevent aggregation (NOTAA FILVWYon surfaces) [95]. - Functional Motif Integration: For enzymes, a pre-defined catalytic motif must be manually grafted into the scaffold and surrounded by a structured protein environment, a process known as "scaffolding" [19].
This entire process is computationally intensive and requires numerous design-test-analyze-redesign cycles to achieve experimental success.
The AI-Driven de novo Design Protocol
Modern AI workflows streamline this process into a more automated and rapid pipeline, as demonstrated by the combination of RFdiffusion and ProteinMPNN [19]:
- Conditioning: The design goal is specified as input to the generative model. For RFdiffusion, this can be unconditional (generating any stable protein) or conditional. Conditioning can include partial structural information (e.g., a fixed functional motif for active site scaffolding), fold topology, or specifications for symmetric oligomers [19].
- Backbone Generation: RFdiffusion generates a novel protein backbone through a denoising process. It starts from random noise (random residue frames) and iteratively refines it over many steps into a coherent, protein-like structure that matches the conditioning input. This process takes only seconds [19].
- Sequence Design: The generated backbone is passed to ProteinMPNN, a neural network that predicts optimal sequences for the structure in a single forward pass. It can generate multiple, diverse sequences that are predicted to fold into the target backbone with high probability [19].
This integrated workflow can generate thousands of candidate proteins—complete with both structure and sequence—in a fraction of the time required for a single Rosetta design.
Performance and Experimental Success
Quantitative benchmarking and experimental validation are critical for assessing the real-world performance of these platforms.
Table 2: Performance and Application Benchmarking of Design Strategies.
| Metric | Rosetta | AI-Driven Workflows (RFdiffusion) |
|---|---|---|
| Design Speed | Hours to days for a single design [50] | ~11 seconds for a 100-residue protein [96] |
| In silico Success Rate | Variable; highly dependent on protocol tuning [95] | >70% of designs are thermostable with expected spectra [96]; high AF2 confidence for monomers [19] |
| Experimental Success (General Folds) | Landmark achievements (e.g., Top7) [50] | High success for symmetric assemblies, binders, monomers [19] |
| Experimental Success (Functional Sites) | Successful for enzyme active site design [50] | Successful scaffolding of functional motifs with atomic accuracy (cryo-EM validation) [19] |
| Key Advantage | Precise, atomic-level control; extensive history of validation [92] | Speed, diversity, and ability to access novel folds beyond the PDB [19] |
The experimental success of AI-driven designs is particularly notable. In one study, hundreds of designs generated by RFdiffusion for symmetric assemblies, metal-binding proteins, and protein binders were experimentally characterized. Many were confirmed to be extremely thermostable, and a cryo-electron microscopy structure of a designed influenza hemagglutinin binder was nearly identical to the design model, confirming atomic-level accuracy [19]. This demonstrates that the AI-generated structures are not just computationally plausible but are also highly designable and expressible in the lab.
Successful de novo enzyme design relies on a suite of computational and experimental tools. The following table details key resources for implementing and validating the discussed workflows.
Table 3: Essential Research Reagents and Resources for De Novo Enzyme Design.
| Item / Resource | Type | Primary Function in Workflow |
|---|---|---|
| Rosetta Software Suite [92] | Software | A comprehensive platform for physics-based macromolecular modeling, docking, and design. |
| RFdiffusion [19] | Software / AI Model | A generative model for creating novel protein backbones conditioned on user specifications. |
| ProteinMPNN [19] | Software / AI Model | A neural network for designing sequences that fold into a given protein backbone structure. |
| AlphaFold2 [50] | Software / Validation Tool | A structure prediction network used for in silico validation of designed protein models. |
| EZSpecificity [97] | Software / AI Tool | Predicts enzyme-substrate specificity, aiding in functional screening of designed enzymes. |
| PyRosetta [92] | Software Interface | A Python-based interactive interface for the Rosetta software suite, enabling scripted protocols. |
| Blueprint File [95] | Input File | A text file defining target secondary structure and loop geometry for Rosetta's de novo protocol. |
| Resfile [95] | Input File | A text file specifying sequence design constraints (allowed/disallowed residues) in Rosetta. |
The comparative analysis reveals that Rosetta and AI-driven workflows are complementary technologies with distinct strengths. Rosetta offers unparalleled, precise control over biomolecular systems, making it ideal for problems where atomic-level engineering is paramount and computational cost is secondary. Its well-established methodology is backed by decades of experimental validation. Conversely, AI-driven workflows like RFdiffusion and ProteinMPNN excel in speed, scalability, and the exploration of novel sequence-structure space. They lower the barrier to entry for de novo design and are particularly powerful for generating diverse backbones and scaffolding functional motifs.
The future of de novo enzyme design lies in the strategic integration of both paradigms. Emerging trends point toward hybrid approaches that leverage the generative power of AI for initial candidate screening and the refining precision of Rosetta for subsequent optimization [94]. Furthermore, the field is moving beyond static structure design toward the incorporation of dynamics and multi-state modeling to create functional enzymes with tunable control and allostery [78] [94]. Community benchmarking challenges, such as the Align Protein Engineering Tournament for PETase design, are crucial for driving progress by providing standardized, real-world tests for these rapidly evolving technologies [98]. As both physics-based and AI-driven methods continue to mature, the de novo design of novel enzyme functions will transition from a formidable challenge to a standard tool in biotechnology and drug development.
The de novo design of novel enzyme functions represents a frontier in synthetic biology and biocatalysis, offering the potential to create catalysts for reactions not found in nature. However, the computational design of a protein scaffold is merely the first step; rigorous experimental validation is crucial to confirm that the designed enzyme not exists but also functions as intended in a biologically relevant environment. This guide details a triad of core experimental techniques—X-ray crystallography, native mass spectrometry (native MS), and in cellulo activity assays—that together provide a comprehensive framework for validating the structure, assembly, and function of de novo designed enzymes. Within the context of a broader research thesis, this multi-faceted approach bridges the gap between in silico models and biologically active catalysts, enabling researchers to debug designs, confirm catalytic mechanisms, and advance therapeutic and industrial applications [5] [99].
The synergy between these methods is particularly powerful. While X-ray crystallography offers an atomic-resolution snapshot of the designed active site, native MS verifies the correct assembly and ligand binding of the enzyme complex under non-denaturing conditions. Finally, in cellulo activity assays confirm that the enzyme performs its intended catalytic function within the complex milieu of a living cell, the ultimate test of a successful design. This technical guide provides detailed methodologies and protocols for each pillar, facilitating their adoption in the workflow of de novo enzyme research.
X-ray Crystallography: Determining Atomic-Level Architecture
X-ray crystallography remains the gold standard for determining the three-dimensional structure of proteins at atomic resolution. For de novo designed enzymes, it is the most direct method to verify that the computationally designed scaffold has folded into the intended conformation and that the active site, including any incorporated abiotic cofactors, is properly formed.
Key Workflow and Standard Samples
Serial crystallography (SX), including serial femtosecond crystallography (SFX) at X-ray free-electron lasers (XFELs) and serial synchrotron crystallography (SSX) at synchrotron sources, has advanced the field by enabling data collection from microcrystals at room temperature, providing insights into native structures and dynamics [100]. A robust crystallization workflow is foundational to this technique.
The diagram below outlines the key steps in a serial crystallography workflow.
For method development and validation, several well-characterized standard proteins are indispensable. The table below summarizes key standard samples used in serial crystallography.
Table 1: Standard Protein Samples for Serial Crystallography Method Validation
| Protein | Molecular Weight | Key Features | Primary Application in SX |
|---|---|---|---|
| Lysozyme | ~14 kDa | Reliable crystallization, high-quality diffraction, compatible with various delivery methods [100]. | Instrument commissioning, method optimization [100]. |
| Glucose Isomerase | 43.3 kDa | Commercial availability, homogeneous microcrystals (diffract to ~2 Å) [100]. | Testing viscous injection matrices, fixed-target setups [100]. |
| Proteinase K | 29.5 kDa | Rapid microcrystal growth (diffract to ~1.8 Å) [100]. | High-speed data acquisition, on-chip crystallization [100]. |
| Myoglobin | ~17 kDa | Photoreactivity, well-defined ligand-binding dynamics [100]. | Time-resolved pump-probe studies [100]. |
| iq-mEmerald | ~27 kDa | Engineered metal sensor, fluorescence modulation upon metal binding [100]. | Visualizing mixing efficiency in time-resolved experiments [100]. |
Application in De Novo Enzyme Design
In de novo enzyme design, crystallography validates critical design features. For instance, in the creation of an artificial metathase, crystallography would be used to confirm the successful supramolecular anchoring of a synthetic Hoveyda-Grubbs catalyst within the designed protein pocket. This would verify the precise orientation of the cofactor and the hydrophobic environment intended to shield the catalytic ruthenium center from cellular nucleophiles like glutathione, which is crucial for in cellulo activity [5].
Native Mass Spectrometry: Probing Stoichiometry and Interactions
Native mass spectrometry (native MS) is a rapidly advancing technique that enables the analysis of intact protein complexes under non-denaturing conditions, preserving non-covalent interactions in the gas phase. It is invaluable for confirming the molecular weight of a de novo designed enzyme, its oligomeric state, and its interaction with substrates, cofactors, or inhibitors.
Technical Workflow and Data Analysis
Native MS involves gently ionizing a protein sample from a volatile buffer and measuring its mass-to-charge ratio. A key application is the identification and characterization of proteoforms—distinct protein species with specific sequences and modifications—using native top-down mass spectrometry (nTDMS) [101].
The precisION software package provides an end-to-end solution for nTDMS data, using a fragment-level open search to discover uncharacterized PTMs and truncations that are critical for understanding the functional form of a designed enzyme [101]. The workflow involves deconvolution of low signal-to-noise spectra, machine learning-based filtering of isotopic envelopes, and hierarchical assignment of fragments, culminating in the identification of modified proteoforms [101].
Key Applications in Hit Validation and Characterization
The ability of native MS to directly observe protein-ligand complexes makes it particularly useful in drug discovery and enzyme design. A prominent application is the hit validation process for DNA-Encoded Library (DEL) technology.
Table 2: Applications of Native MS in Enzyme and Drug Discovery Research
| Application | Description | Utility in De Novo Enzyme Research |
|---|---|---|
| Hit Validation from DEL Selections | Rapidly validates and ranks affinity of "On-DNA" binders without tedious purification steps [102]. | Confirms designed enzymes bind to intended transition-state analogs or inhibitors. |
| Binding Affinity Ranking | Preserves non-covalent interactions, allowing relative affinity determination for a series of ligands [102] [99]. | Quantifies the effect of active site mutations on cofactor or substrate binding. |
| Proteoform-Resolved Characterization | Identifies and quantifies specific protein isoforms with distinct PTMs using nTDMS [101] [99]. | Verifies the correct processing and modification state of a designed enzyme expressed in cells. |
The following diagram illustrates how native MS integrates with the DEL hit validation workflow.
Activity Assays in Cellulo: Confirming Catalytic Function
Validating that a de novo designed enzyme is functional within a living cell is the ultimate test of its design. In cellulo activity assays confirm that the enzyme is stable, properly folded, and catalytically active in the complex cytoplasmic environment, despite potential interference from cellular metabolites, proteases, and the reducing environment.
Designing Robust Enzymatic Assays
A well-designed biochemical assay is the cornerstone of functional validation. The development process typically involves defining the biological objective, selecting a sensitive and reproducible detection method, and rigorously optimizing and validating assay components to ensure robustness, often measured by a Z'-factor > 0.5 for high-throughput screening (HTS) [103].
Universal assay platforms, such as the Transcreener ADP assay for kinases or the AptaFluor SAH assay for methyltransferases, detect common enzymatic products and can be broadly applied across enzyme classes, simplifying the assay development process [103]. These are often homogeneous, "mix-and-read" assays that are amenable to automation and HTS.
Advanced Techniques for Cellular Activity Measurement
Single-Cell Enzyme Activity Assay: For probing cellular heterogeneity, single-time-point stable isotope probing-mass spectrometry (SIP-MS) offers a powerful solution. This method involves delivering a pool of stable isotope-labeled substrate peptides into single cells. After a fixed incubation time, MS simultaneously quantifies the products from all substrate variants, enabling the calculation of reaction rates at different substrate concentrations from a single time point. This approach has been used, for example, to reveal heterogeneity in Cathepsin D activity in breast cancer cells, correlating high activity with increased metastatic potential [104].
Validating Artificial Metalloenzymes in Cells: A demonstrated example involved an artificial metathase for olefin metathesis in the cytoplasm of E. coli. The validation required:
- Directed Evolution: The initial de novo designed protein scaffold was optimized through iterative rounds of mutagenesis and screening in cell-free extracts (CFE) supplemented with copper(II) bisglycinate to oxidize and mitigate the inhibitory effects of glutathione [5].
- Performance Metrics: Evolved variants were characterized by excellent turnover numbers (TON ≥ 1,000) in the cytoplasmic environment, a significant leap from the modest activity of the free cofactor [5].
Integrated Workflow and Research Reagents
In a comprehensive de novo enzyme design project, these techniques are not used in isolation but are integrated into a sequential, iterative validation workflow. The synergy begins with structural validation, proceeds to complex integrity analysis, and culminates in functional testing within the target environment.
The following diagram illustrates how these techniques form a cohesive validation strategy.
Essential Research Reagent Solutions
The experimental workflows described rely on a suite of key reagents and tools. The following table details essential solutions for researchers in this field.
Table 3: Key Research Reagent Solutions for Experimental Validation
| Reagent / Tool | Function | Example Application |
|---|---|---|
| precisION Software | An open-source software package for nTDMS data analysis; performs fragment-level open search to discover hidden PTMs [101]. | Identifying unplanned modifications or truncations in a de novo enzyme expressed in E. coli. |
| Transcreener ADP Assay | A universal, homogeneous immunoassay for detecting ADP formation; applicable to kinases, ATPases, etc [103]. | High-throughput screening of enzyme activity or inhibitor potency for ATP-dependent de novo enzymes. |
| Stable Isotope Labeled Peptides | Serve as multiplexed substrates in SIP-MS assays; allow simultaneous measurement of multiple reaction rates [104]. | Profiling the substrate specificity or kinetic parameters of a designed protease in single cells. |
| Machine Learning Force Fields (MLFFs) | Used in Crystal Structure Prediction (CSP) for accurate energy ranking of polymorphs [105]. | Computational screening of potential crystallization conditions for a de novo enzyme. |
| Abcam CA Activity Kit (ab284550) | Colorimetric kit measuring CA's esterase activity via nitrophenol release [106]. | Standardized benchmarking of carbonic anhydrase activity in surface-display constructs. |
The de novo design of novel enzyme functions is a challenging endeavor that demands rigorous, multi-faceted experimental validation. The integrated use of X-ray crystallography, native mass spectrometry, and in cellulo activity assays provides a powerful framework to conclusively demonstrate that a designed enzyme adopts the intended structure, forms the correct complexes, and performs its catalytic function within the complexity of a living cell. By adopting the detailed protocols and strategies outlined in this guide, researchers can accelerate the design-build-test cycle, debug computational models with empirical data, and confidently advance the field of artificial enzyme design towards new therapeutic and industrial applications.
The de novo design of enzymes represents a grand challenge in computational biology and biotechnology, aiming to create custom biocatalysts for reactions not found in nature. For years, a significant performance gap has separated computationally designed enzymes from their natural counterparts and small-molecule synthetic catalysts. Natural enzymes achieve remarkable catalytic efficiencies, often with kcat/KM values exceeding 10⁵ M⁻¹·s⁻¹ and turnover numbers (kcat) of 10 s⁻¹ or higher [6]. Historically, de novo designed enzymes exhibited efficiencies orders of magnitude lower, typically between 1–420 M⁻¹·s⁻¹ with kcat values well below 1 s⁻¹, necessitating extensive laboratory evolution to bridge this gap [6]. This gap highlighted critical limitations in our understanding of biocatalytic fundamentals and an inability to precisely control all protein degrees of freedom to achieve optimal catalytic constellations.
Recent breakthroughs in computational methodologies, particularly integrating advanced machine learning with atomistic design, are rapidly closing this performance gap. This analysis examines the current state of de novo designer enzymes, directly comparing their catalytic performance, stability, and functional scope against natural catalysts and small-molecule synthetic analogs. We frame this within the broader thesis of de novo design of novel enzyme functions, highlighting the experimental protocols and mechanistic insights that underpin recent successes.
Quantitative Performance Comparison
The catalytic parameters of kcat (turnover number) and kcat/KM (catalytic efficiency) serve as the primary metrics for comparing enzyme performance. The following table summarizes benchmark data for the Kemp elimination reaction, a model reaction for proton abstraction widely used in design studies.
Table 1: Catalytic Performance Metrics for Kemp Elimination Catalysts
| Catalyst Type | Catalytic Efficiency (kcat/KM, M⁻¹·s⁻¹) |
Turnover Number (kcat, s⁻¹) |
Key Characteristics |
|---|---|---|---|
| Natural Eliminases (Median) [6] | ~10⁵ | ~10 | High efficiency, biological relevance |
| Small-Molecule Synthetic Analogs | Varies widely | Varies widely | Tunable chemistry, no protein scaffold |
| Previous Computational Designs (pre-2025) [6] | 1 – 420 | 0.006 – 0.7 | Required intensive experimental optimization |
| Recent Fully Computational Designs (2025) [6] [107] | 2,000 – 12,700 | 0.85 – 2.8 | High stability (>85°C), novel active sites |
| Optimized Fully Computational Design (2025) [6] [107] | >10⁵ | ~30 | Matches natural enzyme parameters |
The data in Table 1 demonstrates a dramatic reduction in the performance gap. The most advanced de novo designs now achieve catalytic parameters comparable to the median values of natural enzymes, a milestone accomplished through fully computational workflows without mutant-library screening [6] [107]. Furthermore, these designs exhibit high thermal stability (over 85°C) and can contain over 140 mutations from any natural protein, featuring novel active sites [107].
Experimental Protocols in De Novo Enzyme Design
The creation of high-efficiency enzymes relies on sophisticated computational and experimental pipelines. The following workflow details a proven, fully computational protocol for designing de novo enzymes in TIM-barrel folds.
Diagram 1: Computational Enzyme Design Workflow
Computational Design Protocol
Step 1: Theozyme Definition
- Objective: Create a quantum-mechanical model of the ideal catalytic constellation for the target reaction.
- Methodology: Use high-level quantum mechanics/molecular mechanics (QM/MM) calculations to derive the transition state (theozyme) geometry [6]. For Kemp elimination, this includes a catalytic base (Asp or Glu) for proton abstraction and an aromatic sidechain for π-stacking with the substrate in the transition state [6]. Unlike earlier approaches, polar interactions stabilizing the transition state can be omitted if they risk lowering the pKa of the catalytic base, relying instead on water molecules [6].
Step 2: Backbone Generation and Stabilization
- Objective: Generate thousands of stable, foldable protein backbones with diverse active-site geometries.
- Methodology:
- Combinatorial Assembly and Design: Generate new backbones by combining fragments from homologous proteins within the TIM-barrel fold [6].
- Stability Optimization: Apply Protein Repair One Stop Shop (PROSS) design calculations to stabilize the designed conformations, ensuring high expressibility and thermal stability [6].
Step 3: Active-Site Design and Optimization
- Objective: Precisely position the theozyme and design the surrounding active site for catalytic competence.
- Methodology:
- Geometric Matching: Use algorithms like RosettaMatch to position the theozyme into each generated backbone [6].
- Sequence Optimization: Use Rosetta atomistic calculations to mutate all active-site positions, optimizing for low system energy and high desolvation of the catalytic base [6].
- Fuzzy-Logic Filtering: Screen millions of designs using an objective function that balances potentially conflicting design goals [6].
Step 4: Final Stabilization
- Objective: Further stabilize the active site and protein core to yield robust, functional enzymes.
- Methodology: Apply methods like FuncLib, which restricts mutations to those found in natural protein diversity or uses atomistic energy as the sole optimization objective for de novo reactions [6]. This can result in designs with over 100 mutations from any natural protein [6].
Experimental Validation Protocol
Step 1: Expression and Folding Analysis
- Objective: Confirm the designed enzymes express solubly and fold correctly.
- Methodology:
- Cloning and Expression: Express the designed genes in a suitable host (e.g., E. coli). Assess expression yield and solubility via SDS-PAGE [6].
- Thermal Denaturation: Use techniques like differential scanning fluorimetry (DSF) to measure thermal stability and confirm cooperative folding, indicated by a single melting transition [6].
Step 2: Catalytic Activity Assay
- Objective: Quantify catalytic efficiency (
kcat/KM) and turnover number (kcat). - Methodology:
- Enzyme Kinetics: For Kemp eliminases, monitor the increase in absorbance at 380–400 nm associated with the formation of the reaction product, 2-cyano-5-nitrophenol [6].
- Parameter Determination: Measure initial reaction rates at varying substrate concentrations. Plot the data and fit to the Michaelis-Menten equation to determine
KMandVmax. CalculatekcatasVmax / [E], where[E]is the enzyme concentration [6].
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents, software, and resources essential for research in de novo enzyme design.
Table 2: Key Research Reagents and Tools for De Novo Enzyme Design
| Item Name | Type | Function / Application |
|---|---|---|
| Rosetta Software Suite | Software | A comprehensive platform for protein structure prediction and design; used for atomistic active-site design and theozyme placement [6]. |
| PROSS (Protein Repair One Stop Shop) | Computational Tool | A method for stabilizing protein structures through computational design, used to enhance the stability of designed backbones [6]. |
| FuncLib | Computational Tool | A method for designing functional protein sites by restricting mutations to evolutionarily allowed amino acids; used for active-site optimization [6]. |
| TIM-barrel Scaffolds | Protein Framework | A highly prevalent and versatile protein fold used as a scaffold for engineering new enzymatic functions due to its favorable active-site cavity [6]. |
| 5-Nitrobenzisoxazole | Chemical Substrate | The benchmark substrate for the Kemp elimination reaction, used to assay the activity of designed Kemp eliminases [6]. |
| Cobalamin (Cbl) Riboswitch | RNA Target | A model system for studying and designing small-molecule interactions with RNA, illustrating principles like base displacement and π-stacking [108]. |
| ESM2 (Evolutionary Scale Modeling) | Protein Language Model | A deep learning model used to generate protein sequences and infer structural and functional information from evolutionary data [34] [109]. |
Underlying Mechanisms and Theoretical Frameworks
The convergence of several advanced theoretical frameworks and a deeper understanding of catalysis has been instrumental in bridging the performance gap.
Key Mechanistic Rules for Design
Recent theoretical work suggests that effective enzymatic function emerges from specific physical constraints and non-equilibrium dynamics. A model built on momentum conservation and dissipative coupling proposes three "golden rules" for the optimal function of a fueled enzyme [45]:
- Friction Matching: The enzyme and the substrate molecule should be attached at the smaller end of each.
- Enzyme Dominance: The conformational change of the enzyme must be comparable to or larger than the conformational change required of the molecule.
- Fast Enzyme Dynamics: The conformational change of the enzyme must be fast enough so that the molecule stretches, rather than just following the enzyme without stretching [45].
These rules move beyond simple energy-barrier crossing models and provide a dynamical paradigm for designing enzymes that efficiently transduce energy.
The Role of AI and Machine Learning
Generative artificial intelligence is revolutionizing enzyme design by enabling the creation of novel sequences and structures conditioned on desired functions [110] [109].
- Contrastive Learning: Frameworks like CLIPzyme use contrastive learning to align representations of enzyme structures and chemical reactions in a shared embedding space. This allows for in silico screening of enzymes for novel reactions by retrieving those with the closest embeddings to a query reaction [34].
- Geometric Deep Learning: Models like EnzymeCAGE (CAtalytic-aware GEometric-enhanced enzyme retrieval model) use Graph Neural Networks (GNNs) to explicitly incorporate the geometric features of the enzyme's catalytic pocket and the reaction centers. This integrates local structural details with global evolutionary information from models like ESM2 for highly accurate function prediction and enzyme retrieval [34].
The relationship between these computational approaches and their application is summarized below.
Diagram 2: Convergence of Methodologies in Enzyme Design
The performance gap between de novo designed enzymes, natural catalysts, and small-molecule analogs is closing rapidly. The integration of robust computational workflows utilizing natural protein fragments, advanced active-site optimization, and generative AI models has enabled the creation of stable, efficient enzymes that catalyze non-natural reactions with parameters rivaling those found in nature. These designer enzymes now achieve catalytic efficiencies (kcat/KM) exceeding 10⁵ M⁻¹·s⁻¹ and turnover numbers (kcat) around 30 s⁻¹, matching the median performance of natural enzymes [6] [107].
This progress, framed within a growing theoretical understanding of mechanochemical coupling in catalysis, signifies a paradigm shift. The field is moving away from reliance on experimental trial-and-error and towards a more predictive, computational discipline. While challenges remain in designing for more complex reactions, the methodologies and tools detailed in this analysis provide a robust foundation for the continued de novo design of novel enzyme functions, with profound implications for sustainable chemistry, therapeutics, and fundamental biological research.
Conclusion
The de novo design of novel enzyme functions represents a paradigm shift in biocatalysis, successfully merging the principles of computational design, artificial intelligence, and directed evolution to create powerful new tools for biomedicine. The integration of these methodologies has enabled the creation of artificial metalloenzymes capable of abiotic catalysis in living cells, such as olefin metathesis, with performance metrics that begin to rival their natural counterparts. Key takeaways include the critical importance of scaffold preorganization, the necessity of robust validation frameworks combining simulation and experiment, and the demonstrated potential of these designer enzymes to operate under challenging industrial and physiological conditions. Looking forward, the continued refinement of AI-driven design tools and a deeper understanding of catalytic mechanisms will pave the way for more sophisticated therapeutic applications. This includes the development of targeted prodrug activation systems, novel enzyme replacement therapies, and the precise manipulation of cellular metabolic pathways, ultimately forging new paths for drug development and personalized medicine.
References
- 1. Enzymes from scratch | The Current [https://news.ucsb.edu/2025/021875/enzymes-scratch]
- 2. Illuminating the universe of enzyme catalysis in the era ... [https://www.sciencedirect.com/science/article/abs/pii/S2405471225002054]
- 3. AI-driven de novo enzyme design: Strategies, applications, ... [https://www.sciencedirect.com/science/article/abs/pii/S0734975025000898]
- 4. Artificial Intelligence-Driven de Novo Design of Robust ... [https://pubmed.ncbi.nlm.nih.gov/41152135/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=pubmed-2&utm_content=1fC8RWxzfM4DxnCcqj9BSTDQRMqkY7xK_afk_xmIBZfqUuhM_d&fc=20240516094352&ff=20251029021957&v=2.18.0.post22+67771e2]
- 5. De novo design and evolution of an artificial metathase for ... [https://www.nature.com/articles/s41929-025-01436-0]
- 6. Complete computational design of high-efficiency Kemp ... [https://www.nature.com/articles/s41586-025-09136-2]
- 7. Enzymes as Targets for Drug Development II - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9964764/]
- 8. Enzyme Miniaturization: Revolutionizing Future Biocatalysts [https://pmc.ncbi.nlm.nih.gov/articles/PMC12226312/]
- 9. How Pharmaceutical Enzymes Are Transforming the APIs ... [https://www.amano-enzyme.com/news/how-pharmaceutical-enzymes-are-transforming-the-apis-industry/]
- 10. Pharmaceutical Enzymes In Drug Development [https://infinitabiotech.com/blog/pharmaceutical-enzymes/]
- 11. Artificial metalloenzyme [https://en.wikipedia.org/wiki/Artificial_metalloenzyme]
- 12. Artificial Metalloenzymes: From Selective Chemical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7411666/]
- 13. Repurposing metalloproteins as mimics of natural ... [https://www.sciencedirect.com/science/article/abs/pii/S0162013421000775]
- 14. Combining chemistry and protein engineering for new-to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8995090/]
- 15. Researchers develop powerful strategy for creating new-to ... [https://chbe.illinois.edu/news/stories/46899]
- 16. Blacksmith Medicines Granted U.S. Patent Covering ... [https://blacksmithmedicines.com/blacksmith-medicines-granted-u-s-patent-covering-antibacterial-compounds-targeting-lpxc/]
- 17. Connecting chemical and protein sequence space to ... [https://www.nature.com/articles/s41586-025-09519-5]
- 18. Blacksmith Medicines Secures European Patent Allowance ... [https://www.prnewswire.com/news-releases/blacksmith-medicines-secures-european-patent-allowance-for-lpxc-targeting-antibacterial-compounds-302624605.html]
- 19. De novo design of protein structure and function with ... [https://www.nature.com/articles/s41586-023-06415-8]
- 20. Short-loop engineering strategy for enhancing enzyme ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11982487/]
- 21. Enzyme Engineering Market Size, Report by 2034 [https://www.precedenceresearch.com/enzyme-engineering-market]
- 22. Generative Artificial Intelligence for Function-Driven De ... [https://www.sciepublish.com/article/pii/701]
- 23. De novo design and evolution of an artificial metathase for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12638248/]
- 24. Rational design of enzyme activity and enantioselectivity - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9902596/]
- 25. De novo design and evolution of an artificial metathase for ... [https://sciety.org/articles/activity/10.1038/s41929-025-01436-0]
- 26. Engineering a Metathesis-Catalyzing Artificial Metalloenzyme ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8154321/]
- 27. Olefin Metathesis [https://chem.libretexts.org/Bookshelves/Inorganic_Chemistry/Supplemental_Modules_and_Websites_(Inorganic_Chemistry)/Catalysis/Catalyst_Examples/Olefin_Metathesis]
- 28. Scripts and files from the de novo design of artificial olefin ... [https://github.com/ikalvet/denovo_metathase_design]
- 29. Computational approaches for rational design of proteins with novel ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3962203/]
- 30. (Part 3). What does this story hold... | Medium De novo enzyme design [https://medium.com/@falk_hoffmann/de-novo-enzyme-design-part-3-0d367c6c9adf]
- 31. Enzymes from scratch [https://www.sciencedaily.com/releases/2025/05/250513150243.htm]
- 32. Sparks of function by de novo protein design - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11366440/]
- 33. Comparison of Three Computational Tools for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11587127/]
- 34. De novo enzyme design (Part 2) [https://medium.com/@falk_hoffmann/de-novo-enzyme-design-part-2-79984a93b6fa]
- 35. CLIPZyme: Reaction-Conditioned Virtual Screening of ... [https://arxiv.org/html/2402.06748v1]
- 36. Enzyme specificity prediction using cross-attention graph ... [https://www.nature.com/articles/s41586-025-09697-2]
- 37. Squidly: Enzyme Catalytic Residue Prediction Harnessing ... [https://elifesciences.org/reviewed-preprints/108186]
- 38. Semantic design of functional de novo genes from a ... [https://www.nature.com/articles/s41586-025-09749-7]
- 39. Discovery, design, and engineering of enzymes based on ... [https://pubmed.ncbi.nlm.nih.gov/40313979/]
- 40. A Technical Guide to Directed Evolution for Enhancing ... [https://www.ranomics.com/a-technical-guide-to-directed-evolution-for-enhancing-protein-stability-and-function]
- 41. Evaluation of machine learning-assisted directed evolution ... [https://www.sciencedirect.com/science/article/abs/pii/S2405471225002200]
- 42. Active learning-assisted directed evolution [https://www.nature.com/articles/s41467-025-55987-8]
- 43. Enabling high-throughput enzyme discovery and ... [https://www.nature.com/articles/s41598-024-64938-0]
- 44. Experimental design for high-throughput screening [https://www.sciencedirect.com/science/article/abs/pii/1359644696100258]
- 45. Mechanistic rules for de novo design of enzymes [https://www.sciencedirect.com/science/article/pii/S2667109325001320]
- 46. A metal-trap tests and refines blueprints to engineer ... [https://www.nature.com/articles/s41467-025-56199-w]
- 47. A bioinformatics approach to design minimal biomimetic ... [https://www.nature.com/articles/s42004-025-01702-z]
- 48. Computationally Guided Design of a Soluble, Abundant ... [https://pubmed.ncbi.nlm.nih.gov/40831097/]
- 49. Integrating Computational Design and Experimental ... [https://www.mdpi.com/2218-273X/14/9/1073]
- 50. The Role of AI-Driven De Novo Protein Design in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12467925/]
- 51. Application of photodynamic activation of prodrugs ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02404-9]
- 52. De Novo-Designed Miniprotein Inhibits the Enzymatic Activity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12570131/]
- 53. Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST) [https://pmc.ncbi.nlm.nih.gov/articles/PMC4457552/]
- 54. De novo design of protein condensation inhibitors by ... [https://www.nature.com/articles/s41467-025-60297-0]
- 55. Tumour-specific STING agonist synthesis via a two- ... [https://www.nature.com/articles/s41557-025-01930-9]
- 56. FDA's New “Plausible Mechanism Pathway” and Other ... [https://www.arnoldporter.com/en/perspectives/advisories/2025/11/fda-plausible-mechanism-pathway-and-other-initiatives-drugs-for-ultra-rare-conditions]
- 57. FDA Outlines “Plausible Mechanism” Approval Pathway for ... [https://www.ropesgray.com/en/insights/alerts/2025/11/fda-outlines-plausible-mechanism-approval-pathway-for-personalized-therapies-but-significant]
- 58. The latest trends in drug discovery [https://www.cas.org/resources/cas-insights/2025-drug-discovery-trends]
- 59. Multi-Timescale Dynamics Study of FKBP12 Along the ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283610011563]
- 60. Possibilities of Using De Novo Design for Generating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9964944/]
- 61. MYC Cofactors: Molecular Switches Controlling Diverse ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4143105/]
- 62. Regulating cellular activity to enhance microbial cell factory ... [https://www.sciencedirect.com/science/article/abs/pii/S0734975025002319]
- 63. [Inactivation and reactivation of proteins (enzymes)] [https://pubmed.ncbi.nlm.nih.gov/6750357/]
- 64. Insight into Coenzyme A cofactor binding and the ... [https://www.nature.com/articles/srep22108]
- 65. Cofactor-independent photo-enzymatic reductions with ... [https://www.nature.com/articles/s41467-025-61908-6]
- 66. Context transcription factors establish cooperative ... [https://www.nature.com/articles/s41588-024-01892-7]
- 67. Data-driven enzyme engineering to identify function ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10365845/]
- 68. Enriching productive mutational paths accelerates enzyme ... [https://www.nature.com/articles/s41589-024-01712-3]
- 69. Enzyme Kinetics Software | KinTek Explorer [https://kintekcorp.com/software]
- 70. ENZO: Enzyme Kinetics [http://enzo.cmm.ki.si/]
- 71. : a naturally occurring tripeptide for functional metal... Glutathione [https://pubs.rsc.org/en/content/articlelanding/2025/sc/d4sc08599j]
- 72. Frontiers | Glutathione : A Samsonian life-sustaining small molecule... [https://www.frontiersin.org/journals/nutrition/articles/10.3389/fnut.2022.1007816/full]
- 73. Glutathione-responsive and -exhausting metal nanomedicines ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10036587/]
- 74. Enhancing the Oral Bioavailability of Glutathione Using ... [https://www.mdpi.com/1999-4923/17/3/385]
- 75. Targeting nanoplatform synergistic glutathione depletion ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11179176/]
- 76. Biocompatibility pathways and mechanisms for bioactive ... [https://www.sciencedirect.com/science/article/pii/S2452199X2100390X]
- 77. Prediction of enzyme function using an interpretable ... [https://www.sciencedirect.com/org/science/article/pii/S2041652025014531]
- 78. De novo protein design—From new structures to ... [https://www.sciencedirect.com/science/article/pii/S0092867423014022]
- 79. Turnover number - Wikipedia [https://en.wikipedia.org/wiki/Turnover_number]
- 80. Turnover Number - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/chemistry/turnover-number]
- 81. Dissociation Constant (KD) Simply Explained [https://fluidic.com/insight/dissociation-constant-kd-simply-explained-and-common-mistakes-to-avoid/]
- 82. What is KD (dissociation constant) [https://www.fidabio.com/knowledge-base/what-is-kd-dissociation-constant]
- 83. Catalytic Efficiency of Enzymes [https://chem.libretexts.org/Bookshelves/Biological_Chemistry/Supplemental_Modules_(Biological_Chemistry)/Enzymes/Enzymatic_Kinetics/Catalytic_Efficiency_of_Enzymes]
- 84. Catalytic Efficiency - an overview [https://www.sciencedirect.com/topics/immunology-and-microbiology/catalytic-efficiency]
- 85. Reflections from Biotrans 2025: Charting the Future of… [https://www.biocatalysts.com/media-resources/biotrans-biocatalysis-future-trends]
- 86. The Empirical Valence Bond as an Effective Strategy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2671572/]
- 87. Why Do Empirical Valence Bond Simulations Yield ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10976637/]
- 88. Advancing predictive modeling in computational chemistry ... [https://link.springer.com/article/10.1007/s44371-025-00363-0]
- 89. researchers develop model to predict enzyme function ... [https://www.fz-juelich.de/en/jsc/news/news-items/news-flashes/2025/juwels-in-action-researchers-develop-model-to-predict-enzyme-function-using-supercomputer]
- 90. Physics-based Modeling in the New Era of Enzyme Engineering [https://pmc.ncbi.nlm.nih.gov/articles/PMC12239909/]
- 91. Enhancing a de novo enzyme activity by computationally- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7407621/]
- 92. Macromolecular modeling and design in Rosetta [https://pmc.ncbi.nlm.nih.gov/articles/PMC7603796/]
- 93. Review Article Artificial intelligence in de novo protein design [https://www.sciencedirect.com/science/article/pii/S2590093525000177]
- 94. From Machine Learning to Multimodal Models: The AI ... [https://www.sciencedirect.com/science/article/pii/S2693125725000457]
- 95. De novo Protein Design [https://forum.rosettacommons.org/node/10002]
- 96. Artificial intelligence-driven approaches for the rational ... [https://www.nature.com/articles/s44431-025-00005-6]
- 97. New AI tool helps match enzymes to substrates [https://las.illinois.edu/news/2025-11-03/new-ai-tool-helps-match-enzymes-substrates]
- 98. A Green Challenge for AI: Engineering Proteins to Tackle Plastic ... [https://alignbio.org/blog/a-green-challenge-for-ai-engineering-proteins-to-tackle-plastic-waste/]
- 99. Traversing the drug discovery landscape using native ... [https://www.sciencedirect.com/science/article/pii/S0959440X25000119]
- 100. Standard Sample Preparation for Serial Femtosecond ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12649929/]
- 101. Uncovering hidden protein modifications with native top ... [https://www.nature.com/articles/s41592-025-02846-5]
- 102. Native Mass Spectrometry Facilitates Hit Validation in DNA ... [https://pubmed.ncbi.nlm.nih.gov/40459511/]
- 103. Biochemical Assay Development: Strategies to Speed Up ... [https://bellbrooklabs.com/accelerate-biochemical-assay-development/?srsltid=AfmBOoqp7D6RjgBEYFXQqNxD_i6vn8cgxiZhaEDmAWMnLUNJ4GzPOYhy]
- 104. Detection of single-cell enzyme activity by single-time-point ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc04171f]
- 105. A robust crystal structure prediction method to support ... [https://www.nature.com/articles/s41467-025-57479-1]
- 106. Functional Validation - 2025 UBC iGEM Wiki [https://2025.igem.wiki/ubc-vancouver/biocementing-bacteria/functional-validation]
- 107. Complete computational design of high-efficiency Kemp ... [https://rosettacommons.org/2025/08/29/complete-computational-design-of-high-efficiency-kemp-elimination-enzymes/]
- 108. Designing small molecules targeting a cryptic RNA binding ... [https://www.nature.com/articles/s41589-025-02018-8]
- 109. Discovery, design, and engineering of enzymes based on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12042125/]
- 110. Generative artificial intelligence for enzyme design [https://www.sciencedirect.com/science/article/pii/S2452223625000148]