Harnessing Light and Enzymes: New Frontiers in Asymmetric Synthesis
This article explores the rapidly evolving field of photoenzymatic enantioselective synthesis, a powerful strategy that merges the energy of visible light with the precise stereocontrol of enzymes.
Harnessing Light and Enzymes: New Frontiers in Asymmetric Synthesis
Abstract
This article explores the rapidly evolving field of photoenzymatic enantioselective synthesis, a powerful strategy that merges the energy of visible light with the precise stereocontrol of enzymes. We delve into the foundational principles of flavin-dependent 'ene'-reductases and other photoenzymes, detailing their mechanism in generating and steering reactive radicals for asymmetric bond formation. The review covers methodological advances in synthesizing valuable chiral building blocks, including fluorinated amides, amines, and diamines, which are pivotal in pharmaceutical and agrochemical development. Practical guidance on troubleshooting reaction parameters and optimizing systems through enzyme engineering is provided. Finally, we present a comparative analysis validating this biocatalytic approach against traditional chemical methods, highlighting its superior sustainability, selectivity, and potential to access novel chemical space for drug discovery.
The Principles of Photoenzymatic Catalysis: Merging Light with Biocatalysis
Defining Photoenzymatic Enantioselective Synthesis
Photoenzymatic enantioselective synthesis represents a cutting-edge interdisciplinary approach that merges the principles of photocatalysis with the precision of enzymatic catalysis to create chiral molecules with high optical purity. This methodology leverages visible light to generate highly reactive radical intermediates through photoexcitation of biological cofactors or integrated photocatalysts, while enzymes provide the chiral environment necessary to control the stereochemical outcome of the reaction. The result is a powerful synthetic platform that achieves exceptional enantioselectivity in transformations that are challenging or impossible to accomplish using traditional chemical or biological methods alone [1].
This hybrid catalytic strategy addresses a fundamental challenge in synthetic chemistry: controlling the stereochemistry of highly reactive, short-lived radical intermediates. By confining these radicals within enzyme active sites, researchers can direct reactions toward single enantiomers with precision that often surpasses what can be achieved with small-molecule chiral catalysts. The field has expanded rapidly in recent years, enabling previously inaccessible asymmetric transformations through the repurposing of natural enzymes for "new-to-nature" reactivities [2]. This approach is particularly valuable for pharmaceutical and agrochemical applications where chiral molecules with defined stereochemistry are essential for biological activity and regulatory approval.
Fundamental Principles and Mechanisms
Photoenzymatic catalysis operates through several distinct mechanistic paradigms, each combining light energy with enzymatic stereocontrol in different configurations:
Net-Reduction Photoenzymatic Catalysis
This common mechanism involves photoinduced electron transfer from reduced enzyme cofactors to substrate molecules, generating radical intermediates that undergo subsequent transformations before being quenched through hydrogen atom transfer. The flavin hydroquinone (FMNH-) in its photoexcited state serves as a potent reductant, capable of directly reducing substrates without requiring external photocatalysts [3]. The enzyme active site then controls the stereochemistry of the radical addition and subsequent hydrogen atom transfer steps.
Redox-Neutral Photoenzymatic Catalysis
Some systems operate without net redox change, utilizing direct visible-light excitation of enzyme-substrate complexes or electron donor-acceptor complexes formed within the enzyme active site. These transformations often involve energy transfer mechanisms or radical pairs generated through homolytic bond cleavage [2].
Synergistic Dual Photo-/Enzymatic Catalysis
In this approach, external photocatalysts (such as transition metal complexes or organic dyes) work cooperatively with enzymes to achieve stereocontrol. The photocatalyst handles the radical generation steps, while the enzyme controls the stereoselectivity of the transformation. This division of labor expands the range of compatible radical precursors beyond those that can be directly activated by biological cofactors [4].
Table 1: Key Photoenzymatic Mechanisms and Their Characteristics
| Mechanistic Type | Radical Generation Method | Stereocontrol Source | Representative Examples |
|---|---|---|---|
| Net-Reduction | Photoexcited flavin hydroquinone (FMNH-) | Enzyme-mediated HAT | Ene-reductase catalyzed hydroalkylation [5] |
| Redox-Neutral | Direct substrate excitation or EDA complexes | Enzyme-controlled radical addition | PLP-dependent radical α-alkylation [6] |
| Dual Catalysis | External photocatalyst (e.g., RhB) | Enzyme active site confinement | ERED-RhB catalyzed hydroamination [4] |
Experimental Protocols
Protocol 1: Photoenzymatic Synthesis of Fluorinated Amides via Ene-Reductase Catalysis
This protocol describes the enantioselective hydroalkylation of alkenes with fluorinated bromoamide radicals using engineered ene-reductases, adapted from published procedures with yields up to 91% and 97% enantiomeric excess (ee) [5].
Reagents and Materials
- Enzyme: OYE1 ene-reductase (wild-type or Y375F mutant)
- Cofactor Regeneration System: Glucose dehydrogenase (GDH, 2 mg/mL), NADP+ (0.5 mM), D-(+)-glucose (50 mM)
- Radical Precursor: Fluorine-containing bromoamide substrate (F-1, 10 mM)
- Alkene Substrate: Electron-rich alkene (15 mM)
- Buffer: Tris-HCl (50 mM, pH 8.5)
- Light Source: Blue LEDs (465 nm, 45 W)
Procedure
- Reaction Setup: In a 5 mL glass vial, combine Tris-HCl buffer (2 mL), fluorinated bromoamide F-1 (0.02 mmol), alkene substrate (0.03 mmol), NADP+ (0.001 mmol), and glucose (0.1 mmol).
- Enzyme Addition: Add OYE1 enzyme (2 mol%) and GDH (2 mg/mL) to the reaction mixture.
- Light Exposure: Seal the vial and irradiate with blue LEDs (465 nm, 45 W) while stirring at 25°C for 24 hours. Maintain constant temperature using a circulating water bath.
- Reaction Monitoring: Monitor reaction progress by regular sampling and GC-MS analysis.
- Product Isolation: After completion, extract the reaction mixture with ethyl acetate (3 × 5 mL). Combine organic layers, dry over anhydrous Na₂SO₄, and concentrate under reduced pressure.
- Purification: Purify the crude product by flash chromatography on silica gel using hexane/ethyl acetate gradient elution.
Analytical Data
Characterize the purified fluorinated amide by ( ^1H ) NMR, ( ^{13}C ) NMR, ( ^{19}F ) NMR, and HRMS. Determine enantiomeric excess by chiral HPLC or GC analysis.
Protocol 2: Intermolecular Radical Hydroamination for Chiral Amine Synthesis
This protocol describes the photoenzymatic generation of nitrogen-centered radicals and their enantioselective addition to olefins using evolved XenB ene-reductase, achieving up to 88% yield and 97% ee [3].
Reagents and Materials
- Enzyme: XenB ene-reductase variant (E2 mutant: XenB-A232L)
- Cofactor Regeneration: GDH (2 mg/mL), NADP+ (0.5 mM), D-glucose (50 mM)
- Nitrogen Radical Precursor: Carbamate with 4-cyanophenolate leaving group (10 mM)
- Olefin Substrate: Styrene derivative (15 mM)
- Buffer: Imidazole-HCl (50 mM, pH 6.5)
- Light Source: Blue LEDs (420-430 nm)
Procedure
- Reaction Setup: In a 5 mL glass vial, combine imidazole-HCl buffer (2 mL), carbamate substrate (0.02 mmol), olefin (0.03 mmol), NADP+ (0.001 mmol), and glucose (0.1 mmol).
- Enzyme Addition: Add XenB variant (2 mol%) and GDH (2 mg/mL) to the reaction mixture.
- Photoreaction: Irradiate with blue LEDs (420-430 nm) while stirring at 25°C for 24-48 hours.
- Reaction Quenching: Extract the reaction mixture with dichloromethane (3 × 5 mL).
- Purification: Combine organic extracts, dry over Na₂SO₄, concentrate, and purify by preparative TLC or flash chromatography.
Analytical Data
Characterize the chiral amine product by ( ^1H ) NMR, ( ^{13}C ) NMR, and HRMS. Determine enantiomeric excess by chiral HPLC after derivatization.
Table 2: Key Reaction Optimization Parameters for Photoenzymatic Systems
| Parameter | Optimal Conditions (Fluorinated Amides) | Optimal Conditions (Hydroamination) | Impact on Yield/Selectivity |
|---|---|---|---|
| Light Source | 465 nm, 45 W blue LEDs | 420-430 nm blue LEDs | Wavelength affects cofactor excitation; power influences radical generation rate |
| Buffer/pH | Tris-HCl, pH 8.5 | Imidazole-HCl, pH 6.5 | pH affects cofactor redox potential and substrate binding |
| Enzyme Loading | 2 mol% OYE1 | 2 mol% XenB variant | Higher loading increases reaction rate but may cause scattering |
| Cofactor System | GDH/glucose/NADP+ | GDH/glucose/NADP+ | Maintains reduced flavin pool for continuous radical generation |
| Reaction Time | 24 hours | 24-48 hours | Longer times needed for electron-deficient substrates |
Visualization of Experimental Workflows
General Photoenzymatic Catalysis Workflow
Ene-Reductase Catalyzed Fluorinated Amide Synthesis
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Photoenzymatic Enantioselective Synthesis
| Reagent/Enzyme | Function/Role | Application Examples | Optimization Tips |
|---|---|---|---|
| Ene-Reductases (OYE1, XenB) | Flavin-dependent enzymes for radical generation & stereocontrol | Hydroalkylation, hydroamination, lactonization | Enzyme engineering improves activity & selectivity [5] [3] |
| Glucose Dehydrogenase (GDH) | Cofactor regeneration system | Maintains NADPH pool for continuous flavin reduction | Use excess GDH/glucose to ensure cofactor turnover |
| Flavin Mononucleotide (FMN) | Native photoactive cofactor | Electron transfer, radical generation | Ensure sufficient FMN loading (0.5-1 mol%) |
| Naphthyl-Substituted Amides | Fluorinated radical precursors | Synthesis of chiral fluorinated pharmaceuticals | Amide group enhances enzyme binding vs. esters [5] |
| Katritzky Pyridinium Salts | Electrophilic radical precursors | α-Alkylation of amino acids | Enable redox-neutral transformations [6] |
| Rhodamine B | External organic photocatalyst | Synergistic catalysis with EREDs | Enables green light excitation (530-540 nm) [4] |
| N-Amido Pyridinium Salts | Nitrogen-centered radical precursors | Vicinal diamine synthesis | N-N bond cleavage under mild conditions [4] |
Applications in Pharmaceutical Synthesis
Photoenzymatic enantioselective synthesis has enabled efficient routes to valuable pharmaceutical intermediates and active ingredients:
Fluorinated Pharmaceutical Compounds
The incorporation of fluorine atoms into drug molecules enhances metabolic stability, membrane permeability, and binding affinity. The photoenzymatic synthesis of fluorinated amides with distal chirality provides access to structural motifs found in numerous pharmaceuticals [5]. This method addresses the challenge of stereoselective fluorination, which is difficult to achieve using conventional synthetic approaches.
Chiral Amine Synthesis
Chiral amines constitute key structural elements in approximately 40% of small-molecule pharmaceuticals. The photoenzymatic hydroamination platform enables direct enantioselective incorporation of amine groups into olefin scaffolds, providing streamlined access to these valuable building blocks [3]. This approach is particularly valuable for creating amine-containing stereocenters remote from functional groups that would typically anchor substrates to traditional catalysts.
Non-Canonical Amino Acids
Photoenzymatic methods have enabled the synthesis of challenging α-tri- and tetrasubstituted non-canonical amino acids (ncAAs) using PLP-dependent threonine aldolases [6]. These unnatural amino acids serve as essential building blocks for peptide therapeutics and bioactive natural products, with applications in drug development and chemical biology.
Troubleshooting and Optimization Guidelines
Successful implementation of photoenzymatic reactions requires careful attention to several critical parameters:
Light Source Optimization
- Wavelength Selection: Match light wavelength to the absorption maxima of the photoredox cofactor (typically 440-470 nm for flavins).
- Intensity Control: Optimize light intensity to balance radical generation rate with potential enzyme photodamage.
- Uniform Illumination: Ensure reaction vessels are positioned to receive even light distribution for consistent results.
Enzyme Engineering Strategies
- Active Site Mutagenesis: Target residues surrounding the substrate binding pocket to improve activity and selectivity.
- Directed Evolution: Employ iterative rounds of mutagenesis and screening to optimize enzyme performance for non-natural reactions.
- Cofactor Engineering: Modify cofactor binding pockets to alter redox properties or accommodate synthetic photocatalysts.
Reaction Condition Optimization
- Buffer Composition: Screen different buffer systems and pH values, as these significantly impact both enzyme activity and radical stability.
- Cofactor Regeneration: Ensure robust NADPH regeneration systems to maintain reduced flavin pools throughout the reaction.
- Substrate Engineering: Design radical precursors with appropriate redox potentials and leaving groups for efficient activation by photoexcited cofactors.
The protocols and guidelines presented here provide a foundation for implementing photoenzymatic enantioselective synthesis in research settings. As the field continues to evolve, these methodologies are expected to expand the toolbox available for asymmetric synthesis, particularly for challenging transformations involving radical intermediates.
Ene-reductases (EREDs) from the Old Yellow Enzyme (OYE) family are flavin-dependent biocatalysts that have become indispensable tools for asymmetric organic synthesis. These enzymes traditionally catalyze the stereoselective hydrogenation of activated alkenes, a pivotal transformation for generating chiral synthons with up to two stereogenic centers [7]. The versatility of these enzymes is profoundly expanded when combined with light, enabling novel radical reactions that are challenging to achieve with traditional small-molecule catalysts. This application note details the core components of these systems—the ene-reductase protein scaffolds and their flavin mononucleotide (FMN) cofactors—framed within contemporary research on photoenzymatic enantioselective synthesis. It provides a structured overview of their catalytic capabilities, quantitative performance data, and detailed protocols for implementing both traditional and photochemical enantioselective reactions.
Core Components and Mechanisms
The Ene-Reductase Enzyme Scaffold
Ene-reductases are flavoproteins that belong to the Old Yellow Enzyme family, first discovered over 80 years ago [7]. They are ubiquitous in nature, found in yeasts, bacteria, plants, and parasitic eukaryotes [8]. While their physiological roles are not fully elucidated, they are suggested to participate in detoxification processes, response to oxidative stress, and specialized metabolic pathways such as jasmonic acid biosynthesis in plants and prostaglandin biosynthesis in trypanosomes [8].
The discovery and characterization of new EREDs continue to expand the biocatalytic toolbox. For instance, the thermophilic ene-reductase CaOYE from Chloroflexus aggregans demonstrates the potential of exploring extremophile microorganisms. CaOYE exhibits high thermostability, good solvent tolerance, and a wide pH optimum, making it a robust biocatalyst for potentially harsh industrial conditions [8]. Structurally, EREDs are known to assemble as dimers, though CaOYE behaves as a monomer in solution, highlighting the diversity within this enzyme family [8].
The Flavin Cofactor: A Versatile Redox Agent
The catalytic prowess of EREDs is derived from their non-covalently bound flavin mononucleotide (FMN) cofactor. The flavin ring system is a versatile redox-active chromophore that can exist in three stable states: oxidized (FMNox), semiquinone (FMNsq, a radical species), and hydroquinone (FMNhq) [9] [10].
The traditional catalytic cycle for alkene reduction involves a hydride transfer from FMNhq to the β-carbon of an activated alkene substrate, followed by protonation of the resulting enolate by a conserved active-site tyrosine residue [9]. This concerted mechanism yields the saturated product and returns the flavin to its oxidized state (FMNox). Regeneration of FMNhq is typically achieved using NAD(P)H as a stoichiometric hydride donor [7].
Beyond this ground-state chemistry, the flavin cofactor can undergo photoexcitation with visible light. The excited state of the reduced flavin hydroquinone (FMNhq*) is an exceptionally potent single-electron reductant, with a reduction potential estimated at -2.26 V vs. SCE, enabling the activation of challenging radical precursors such as chlorinated amides [9]. This photochemical mechanism mirrors processes in natural photoenzymes like DNA photolyase and forms the basis for non-natural radical reactions catalyzed by engineered EREDs [9].
Table 1: Key States of the Flavin Cofactor and Their Roles
| Flavin State | Redox Potential (vs. SCE) | Primary Role in Catalysis |
|---|---|---|
| Oxidized (FMNox) | — | Electron acceptor in reductive cycles |
| Hydroquinone (FMNhq) | -215 mV | Hydride donor in ground-state alkene reduction |
| Excited Hydroquinone (FMNhq*) | -2.26 V | Potent single-electron reductant for radical initiation |
| Semiquinone (FMNsq) | — | Hydrogen atom donor for radical termination |
Quantitative Performance Data
The performance of ene-reductases is quantified through metrics such as turnover number, enantioselectivity, and stability. Engineered photoenzymes demonstrate remarkable efficiency in novel transformations.
Table 2: Quantitative Performance of Selected Ene-Reductase Catalyzed Reactions
| Enzyme / System | Reaction Type | Typical Yield | Stereoselectivity | Turnover Number (kcat)/Efficiency |
|---|---|---|---|---|
| VEnT1.3 Photoenzyme [11] | Intramolecular [2+2] Cycloaddition | Quantitative (>99%) | >99% e.e. | ( k_{cat} = 13.0 \text{ s}^{-1} ); >1,300 total turnovers |
| NCR-C9 ERED [9] | Radical Hydrodehalogenation & Cyclization | Up to 92% | Up to 97:3 e.r. | — |
| GluER-T36A ERED [9] | Photochemical Radical Cyclization | Up to 92% | 94:6 e.r. | — |
| CaOYE [8] | Traditional Alkene Reduction | Variable, substrate-dependent | — | High thermostability & solvent tolerance |
Experimental Protocols
Protocol 1: Photoenzymatic Intramolecular [2+2] Cycloaddition
This protocol describes a semipreparative-scale enantioselective cycloaddition using the engineered photoenzyme VEnT1.3, which contains a genetically encoded thioxanthone sensitizer [11].
Reagents:
- Photoenzyme: Purified VEnT1.3 (0.5 mol%) [11].
- Substrate: Carbon-linked quinolone derivative
1. - Buffer: Aerobic phosphate or Tris buffer (50-100 mM, pH 7.5-8.0).
- Light Source: High-power LED lamp (λ = 405 nm).
Procedure:
- Reaction Setup: In a glass vial, dissolve substrate
1(e.g., 0.1 mmol) in the appropriate buffer to a final concentration of 5-10 mM. Add the VEnT1.3 photoenzyme to a final loading of 0.5 mol%. - Irradiation: Seal the vial and place it under the 405 nm LED light source with constant stirring. Maintain the reaction temperature at 25-30°C using a water bath or cooling fan.
- Reaction Monitoring: Monitor reaction progress by analytical HPLC or TLC. The reaction typically reaches >99% conversion within minutes, depending on light intensity [11].
- Work-up: Upon completion, extract the reaction mixture with an organic solvent (e.g., ethyl acetate). Combine the organic layers, dry over anhydrous MgSO₄, and concentrate under reduced pressure.
- Product Purification: Purify the crude product (
(+)-1a) using flash chromatography on silica gel. Product identity and enantiopurity can be confirmed by ( ^1\text{H} ) NMR and chiral HPLC, respectively [11].
- Reaction Setup: In a glass vial, dissolve substrate
Protocol 2: ERED-Catalyzed Asymmetric Radical Cyclization
This protocol outlines the setup for an enantioselective radical cyclization using an engineered ERED (e.g., NCR-C9 or GluER-T36A) under photochemical conditions [9].
Reagents:
- Enzyme: Purified ERED variant (e.g., NCR-C9 for bromoketones, GluER-T36A for chloroamides).
- Cofactor Recycling System: NADP⁺ (catalytic amount), engineered glucose dehydrogenase (GDH-105), and D-glucose.
- Substrate: Radical precursor (e.g., α-bromoketone
5or α-chloroamide10). - Buffer: Phosphate buffer (50-100 mM, pH 7.0-8.0).
- Light Source: Violet or cyan LED lamp (λ = 400-500 nm, selected based on the substrate-enzyme pair).
Procedure:
- Cofactor System Preparation: In a reaction vial, combine the buffer, NADP⁺ (0.1-1 mol%), GDH-105 (1-5 mg), and an excess of D-glucose (e.g., 5-10 equiv).
- Reaction Initiation: Add the ERED catalyst and the radical precursor substrate. Seal the vial.
- Irradiation and Mixing: Place the vial under the appropriate LED light source with vigorous stirring to ensure oxygenation for FMN reduction. Irradiate for the specified duration (typically several hours).
- Reaction Monitoring: Monitor consumption of the starting material by TLC or GC-MS.
- Work-up and Analysis: Extract the product with an organic solvent, concentrate the organic phase, and purify the product via flash chromatography. Determine enantiomeric ratio (e.r.) by chiral HPLC or GC [9].
Diagram 1: Photochemical Radical Cyclization Workflow.
The Scientist's Toolkit: Key Research Reagent Solutions
Successful implementation of ene-reductase catalysis, especially for non-natural photochemical reactions, relies on key reagents and materials.
Table 3: Essential Research Reagents for Ene-Reductase Applications
| Reagent / Material | Function / Role | Example Use Case |
|---|---|---|
| Engineered EREDs (NCR-C9, GluER) | Chiral scaffold for enantioselective radical termination and substrate binding. | Asymmetric radical cyclization & hydrodehalogenation [9]. |
| Glucose Dehydrogenase (GDH-105) | Cofactor recycling enzyme; regenerates NADPH from NADP+ using glucose. | Sustained catalytic cycles in radical reactions [9]. |
| Flavin Mononucleotide (FMN) | Redox-active cofactor; core photocatalyst in electron transfer reactions. | Required for in vitro reconstitution of ERED activity. |
| NADP⁺ | Oxidized nicotinamide coenzyme; electron acceptor in GDH-catalyzed recycling. | Catalytic NADPH regeneration in photobiocatalytic systems [9]. |
| Visible Light LEDs (400-500 nm) | Energy source for flavin photoexcitation to access potent redox states. | Enabling radical reactions with low reduction potential substrates [9]. |
| Thioxanthone Photoenzyme (VEnT1.3) | Genetically encoded triplet sensitizer for visible light-driven energy transfer. | Enantioselective [2+2] cycloadditions under visible light [11]. |
Signaling Pathways and Workflow Visualization
The catalytic mechanisms of ene-reductases, both traditional and photochemical, can be visualized as interconnected pathways. The diagram below integrates the hydride transfer, ground-state electron transfer, and photoinduced electron transfer pathways, highlighting the central role of the flavin cofactor.
Diagram 2: Ene-Reductase Catalytic Pathways.
The merger of photocatalysis with enzymatic catalysis represents a paradigm shift in synthetic chemistry, opening avenues for enantioselective transformations that are challenging to achieve through conventional methods. This field leverages light to generate high-energy radical intermediates via photoinduced electron transfer (PET), while enzymes exert exquisite stereochemical control over these reactive species. The integration of these two powerful strategies enables access to new-to-nature enzymatic functions and provides sustainable routes for synthesizing complex chiral molecules, including pharmaceuticals and agrochemicals. This article delves into the mechanistic principles underpinning radical generation through PET and provides detailed protocols for implementing these reactions within photoenzymatic frameworks.
Core Mechanistic Pathways and Quantitative Data
Photoenzymatic catalysis utilizes light energy to initiate single-electron transfer events, generating radical intermediates that are subsequently managed within enzyme active sites. The following table summarizes key catalytic systems and their performance characteristics.
Table 1: Representative Photoenzymatic Systems for Enantioselective Synthesis
| Catalytic System | Reaction Type | Key Radical Intermediate | Performance (Yield / e.e.) | Primary Light Source |
|---|---|---|---|---|
| Ene-Reductase (ERED) + Photocatalyst [12] | Intermolecular Hydroalkylation of Alkenes | Prochiral Carbon-centered Radical | Up to 99% yield, 99% e.e. [12] | Visible Light |
| Ene-Reductase (Direct Flavin Excitation) [13] | Redox-Neutral Hydroarylation | Carbon-centered Radical from Alkene | Excellent enantioselectivity reported [13] | Visible Light |
| Synergistic ERED & Rhodamine B [14] | Hydroamination of Enamides | Nitrogen-Centered Radical (NCR) | >99% e.e. [14] | Green Light (530-540 nm) |
| Flavin-dependent 'Ene'-Reductase [15] | Hydrocyanoalkylation of Alkenes | Cyanoalkyl Radical | High enantioselectivity [15] | Visible Light |
The mechanism of action across different systems hinges on the precise initiation of radical species. Two primary pathways have been established:
- Enzyme-Photocatalyst Synergy: An external photoredox catalyst absorbs light to initiate the radical reaction, while a separate enzyme controls stereoselectivity. For example, in the hydrocyanoalkylation of alkenes, the flavin hydroquinone (FMNhq) within ene-reductases can singly reduce a cyanoalkyl radical precursor. This leads to the formation of a cyanoalkyl radical, which adds across an alkene. The enzyme's active site then stereoselectively controls the radical-terminating hydrogen atom transfer (HAT). [15]
- Direct Flavin Excitation: In systems without an external photocatalyst, the enzyme's native flavin cofactor (FMN or FAD) serves a dual role. Upon photoexcitation, the flavin can directly participate in single-electron transfer to or from a substrate, generating a radical pair. This mechanism is exemplified in the redox-neutral hydroarylation of alkenes, where photoexcited flavin semiquinone (FMNsq) acts as a single-electron oxidant. [13]
Experimental Protocols
Protocol: Intermolecular Radical Hydroalkylation via Photoenzymatic Catalysis
This protocol describes the enantioselective intermolecular hydroalkylation of terminal alkenes with α-halo carbonyl compounds, catalyzed by a flavin-dependent 'ene'-reductase, adapted from published procedures. [12]
Principle: Visible light irradiation triggers an electron transfer that generates a radical from an α-halo carbonyl compound. This radical adds to a terminal alkene, and the resulting prochiral radical intermediate is stereoselectively reduced within the enzyme's active site.
Materials:
- Enzyme: Flavoprotein 'ene'-reductase (e.g., GluER variant [14]).
- Cofactor: NADP+ (1.5 mol%).
- Substrates: Terminal alkene (e.g., α-methylstyrene), α-halo carbonyl compound (e.g., α-bromoamide).
- Cofactor Regeneration System: Glucose dehydrogenase (GDH, 4 U), D-Glucose.
- Solvent: Phosphate buffer (KPi, 100 mM, pH 6.0) with 20% v/v DMSO.
- Light Source: Blue LEDs (e.g., 390-400 nm or 450-455 nm) or Green LEDs (530-540 nm [14]).
- Reaction Vessel: Schlenk tube or vial suitable for irradiation under inert atmosphere.
Procedure:
- Reaction Setup: In a 2.0 mL reaction vial, sequentially add:
- KPi buffer (100 mM, pH 6.0) containing 20% v/v DMSO.
- Terminal alkene (0.008 mmol).
- α-Halo carbonyl compound (0.032 mmol, 4.0 equiv).
- 'Ene'-reductase (2 mol%).
- NADP+ (1.5 mol%).
- Glucose dehydrogenase (4 U).
- D-Glucose (0.024 mmol).
- Degassing: Purge the reaction mixture with nitrogen (N₂) for 10-15 minutes to create an anaerobic environment.
- Irradiation: Place the sealed reaction vessel approximately 5 cm from the LED light source and stir at room temperature for 16 hours.
- Reaction Monitoring: Monitor reaction progress by TLC or LC-MS.
- Work-up: After completion, extract the reaction mixture with ethyl acetate (3 × 2 mL). Combine the organic layers, dry over anhydrous Na₂SO₄, and concentrate under reduced pressure.
- Purification: Purify the crude product by flash column chromatography on silica gel to obtain the desired chiral γ-stereogenic carbonyl compound.
- Analysis: Determine enantiomeric excess (e.e.) by chiral HPLC or SFC analysis.
Troubleshooting Tips:
- Low Conversion: Ensure the light source is of sufficient intensity and that the reaction vessel is transparent to the relevant wavelengths. Check enzyme activity and the integrity of the cofactor regeneration system.
- Poor Enantioselectivity: Screen different 'ene'-reductase homologues or mutants, as the active site architecture is critical for stereocontrol. [12] [14]
Protocol: Hydroamination via Synergive Photo/Biocatalysis
This protocol outlines the enantioselective synthesis of vicinal diamines via hydroamination of enamides, using a dual system of an ene-reductase and an organophotocatalyst. [14]
Principle: Green light excites the organophotocatalyst Rhodamine B (RhB), which engages in single-electron transfer with an N-amidopyridinium salt. This triggers N–N bond cleavage to generate a nitrogen-centered radical (NCR). The NCR adds to the enamide, and the resulting radical intermediate is stereoselectively reduced by the flavin hydroquinone within the ene-reductase.
Materials:
- Biocatalyst: Ene-reductase mutant (e.g., GluER-M5 or GluER-Y177F-F269V). [14]
- Photocatalyst: Rhodamine B (RhB).
- Substrates: Enamide, N-amidopyridinium salt.
- Cofactor System: NADP+, Glucose Dehydrogenase (GDH), D-Glucose.
- Solvent: KPi buffer (100 mM, pH 6.0) with 20% v/v DMSO.
- Light Source: Green LEDs (530-540 nm).
Procedure:
- Reaction Setup: In a 2.0 mL vial, combine:
- Enamide 1 (0.008 mmol).
- N-amidopyridinium salt 2 (0.032 mmol, 4.0 equiv).
- GluER mutant (2 mol%).
- GDH (4 U).
- NADP+ (1.5 mol%).
- Glucose (0.024 mmol).
- Rhodamine B (catalytic amount).
- KPi buffer with 20% v/v DMSO to a total volume of 2.0 mL.
- Degassing: Sparge the mixture with N₂ for 10 minutes.
- Irradiation: Stir the reaction under irradiation with green LEDs for 16 hours at room temperature.
- Work-up and Purification: Extract with ethyl acetate, dry the organic phase, and concentrate. Purify the residue by preparative TLC or column chromatography.
- Analysis: Determine yield and enantiomeric excess as previously described.
Signaling Pathway and Workflow Visualization
The following diagram illustrates the general catalytic cycle for a synergistic photoenzyme system, integrating the key steps of photoinduced radical generation and enzymatic stereocontrol.
Diagram 1: Synergistic Photoenzyme Catalytic Cycle. This workflow shows the integration of photochemical radical generation with enzymatic stereocontrol, typical for hydroalkylation and hydroamination reactions. HAT: Hydrogen Atom Transfer.
The mechanistic pathway for direct flavin excitation, an alternative to synergistic systems, is shown below.
Diagram 2: Direct Flavin Excitation Pathway. This mechanism involves direct photoexcitation of the enzyme-bound flavin cofactor, which acts as an internal photoredox catalyst for single-electron oxidation. SET: Single-Electron Transfer.
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of photoenzymatic radical reactions requires careful selection of components. The following table lists essential reagents and their functions.
Table 2: Key Reagents for Photoenzymatic Radical Generation and Control
| Reagent / Component | Function / Role | Specific Examples |
|---|---|---|
| Flavin-Dependent 'Ene'-Reductases | Biocatalyst for stereocontrol; provides flavin cofactor (FMN) as inherent photoredox center. | GluER variants (e.g., GluER-M5, GluER-Y177F-F269V) [14] [15] |
| Organophotoredox Catalysts | External photosensitizer to absorb light and drive electron transfer reactions. | Rhodamine B (RhB) [14] |
| Radical Precursors | Stable molecules that, upon single-electron reduction or oxidation, fragment to yield reactive radicals. | α-Halo carbonyl compounds [12], N-Amidopyridinium salts [14] |
| Cofactor Regeneration System | Maintains the required redox state of enzymatic cofactors (e.g., NADPH for FMN reduction) sustainably. | Glucose Dehydrogenase (GDH) / Glucose [14] |
| Electron Shuttles (in bio-hybrid systems) | Mediate electron transfer from microbial metabolism to chemical reactants under light excitation. | Endogenously secreted Flavins (Riboflavin, FMN) [16] |
Understanding Electron Donor-Acceptor (EDA) Complex Formation
Electron Donor-Acceptor (EDA) complexes, also referred to as charge-transfer complexes, represent a pivotal class of supramolecular assemblies formed through the association of electron-rich (donor) and electron-deficient (acceptor) molecules [17]. These complexes are characterized by ground-state electronic interactions that enable new absorption bands in the visible light spectrum. Upon photoexcitation, this interaction facilitates a single electron transfer (SET) from the donor to the acceptor, generating highly reactive radical pairs capable of initiating unique transformations under mild conditions [18].
Within the rapidly evolving field of photoenzymatic enantioselective synthesis, EDA complex photochemistry has emerged as a powerful strategy to complement and enhance biocatalytic reactivity. This approach enables the generation of non-natural radical intermediates, including challenging nitrogen-centered radicals (NCRs) and carbon-centered radicals, which are then precisely orchestrated within the enzyme's chiral active site to produce enantioenriched molecules [19] [4]. The integration of EDA complexes with enzymes addresses long-standing challenges in controlling the reactivity and selectivity of high-energy radical species, opening new synthetic pathways for pharmaceutical and bioactive molecule development.
Fundamental Principles and Quantitative Characterization
Mechanistic Workflow of EDA Complex Photoactivation
The following diagram illustrates the generalized mechanism of EDA complex formation, photoexcitation, and subsequent radical generation that can be coupled with enzymatic stereocontrol.
Quantitative Characterization of EDA Complexes
The formation and stability of EDA complexes can be quantitatively monitored using spectroscopic techniques, particularly UV-Vis absorption spectroscopy. The following table summarizes key characterization parameters for a representative EDA complex formed between 1,2-dinitrobenzene (acceptor) and tri-n-butylamine (donor) [20].
Table 1: Quantitative Characterization of a Model EDA Complex
| Parameter | Experimental Value | Experimental Conditions | Significance |
|---|---|---|---|
| Charge-Transfer Band (λ_max) | 490 nm | Acetonitrile, equimolar donor/acceptor | Confirms visible light absorption enabling photoexcitation |
| Optimal Binding Stoichiometry | 4:1 (Acceptor:Donor) | Job's Plot method | Deviates from common 1:1 ratio, informs reaction stoichiometry |
| Solvent Effect | Significant absorption reduction | Addition of 600 μL water to acetonitrile solution | Hydrogen-bonding solvents can disrupt EDA complex formation |
| Molar Absorptivity | Concentration-dependent increase | Constant acceptor, increasing donor | Validates complex formation and allows determination of association constant |
Experimental Protocols for EDA Complex Studies
Protocol 1: Spectroscopic Identification and Characterization
This protocol describes the procedure for identifying EDA complex formation between an electron acceptor and donor using UV-Vis spectroscopy [20].
Objective: To confirm and characterize EDA complex formation between 1,2-dinitrobenzene (acceptor) and tri-n-butylamine (donor) in acetonitrile.
Materials:
- Acceptor Solution: 1,2-Dinitrobenzene (0.01 M in acetonitrile). Note: Purify via column chromatography for pale-yellow pure reagent.
- Donor Solution: Tri-n-butylamine (0.01 M in acetonitrile).
- Solvent Control: HPLC-grade acetonitrile.
- Equipment: UV-Vis spectrophotometer, 1 cm quartz cuvettes, adjustable-volume micropipettes.
Procedure:
- Baseline Correction: Place acetonitrile in both sample and reference cuvettes and record a baseline spectrum from 800 nm to 350 nm.
- Individual Component Scans:
- Scan the acceptor solution (1,2-dinitrobenzene) against acetonitrile reference.
- Scan the donor solution (tri-n-butylamine) against acetonitrile reference.
- EDA Complex Formation:
- Prepare a solution containing equimolar amounts (e.g., 1 mL each) of donor and acceptor solutions in a clean vial.
- Incubate for 1 minute at room temperature.
- Transfer the mixture to a cuvette and scan against acetonitrile reference.
- Stoichiometry Determination (Job's Plot):
- Prepare a series of solutions with varying donor:acceptor ratios while keeping the total concentration constant.
- Measure absorbance at 490 nm for each ratio.
- Plot absorbance versus mole fraction of acceptor to determine optimal binding stoichiometry.
- Solvent Interference Test:
- Add 600 μL of water to the EDA complex solution, mix thoroughly, and record the spectrum.
Expected Outcomes: A successful experiment will show a significant bathochromic shift (red shift) and increased absorption in the visible region (400-500 nm) for the mixture compared to individual components, confirming EDA complex formation. Water addition should substantially decrease absorption intensity.
Protocol 2: Photoenzymatic Hydroamination via EDA Complex
This protocol describes a specific application of EDA complexes in photoenzymatic synthesis for the enantioselective synthesis of vicinal diamines, adapted from recent literature [4].
Objective: To achieve enantioselective hydroamination of enamides using a dual bio-/photo-catalytic system that generates N-centered radicals via EDA complex photoactivation.
Materials:
- Enzyme: GluER-T36A-Y177F-F269V-K277M-Y343F mutant (ER-M5, 1 mol%)
- Cofactor System: Glucose dehydrogenase (GDH), NADP+, D-glucose
- Photocatalyst: Rhodamine B (RhB)
- Substrates: N-(1-phenylvinyl)acetamide (1a), N-amidopyridinium salt (2a)
- Solvent: Phosphate buffer (50 mM, pH 7.5)/acetonitrile (4:1)
- Light Source: Green LEDs (530-540 nm)
- Equipment: Schlenk tube, LED photoreactor, inert atmosphere capability
Procedure:
- Reaction Setup:
- In a dried Schlenk tube, add N-(1-phenylvinyl)acetamide (0.1 mmol) and N-amidopyridinium salt (0.15 mmol).
- Add Rhodamine B (2 mol%) and the ER-M5 enzyme (1 mol%).
- Add the cofactor system: NADP+ (0.5 mM), GDH (2 mg), D-glucose (10 equiv).
- Add phosphate buffer/acetonitrile solvent mixture (2 mL total volume).
- Degassing:
- Freeze the reaction mixture using liquid N₂.
- Evacuate the Schlenk tube under high vacuum.
- Backfill with argon.
- Repeat freeze-pump-thaw cycle three times.
- Photoreaction:
- Place the Schlenk tube in a green LED photoreactor (530-540 nm).
- Stir vigorously at 25°C for 24-48 hours.
- Reaction Monitoring:
- Monitor reaction progress by TLC or LC-MS.
- Determine enantiomeric excess by chiral HPLC or SFC after purification.
- Work-up and Purification:
- Extract the reaction mixture with ethyl acetate (3 × 5 mL).
- Combine organic layers, dry over anhydrous Na₂SO₄, and concentrate.
- Purify the crude product by flash column chromatography.
- Reaction Setup:
Expected Outcomes: The reaction should provide enantioenriched vicinal diamine products with excellent enantioselectivity (up to >99% ee) and good yields (up to 82%). Control experiments should confirm the essential role of light, enzyme, and the EDA complex in the transformation.
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and materials essential for experimental work with EDA complexes in photoenzymatic synthesis.
Table 2: Essential Research Reagents for EDA Complex Photoenzymatic Studies
| Reagent/Material | Function/Application | Representative Examples |
|---|---|---|
| Electron Acceptors | Forms EDA complex with electron donors; generates radical anions upon SET | Aryl halides [18], Katritzky salts [18], 1,2-dinitrobenzene [20], nitroaromatics [20] |
| Electron Donors | Provides electrons in EDA complex; generates radical cations upon SET | Thiophenols [18], tertiary amines [20], enolates [19], dithiocarbamate anions [18] |
| Enzyme Biocatalysts | Provides chiral environment for enantioselective radical transformation | Ene-reductases (EREDs) [19] [4], flavin-dependent 'ene'-reductases [19], GluER mutants [4] |
| Cofactor Systems | Regenerates reduced enzyme form for catalytic turnover | Glucose/GDH/NADP+ [4], flavin mononucleotide (FMN) [19] |
| Photoredox Catalysts | Enhances radical generation in synergistic systems; absorbs specific wavelengths | Rhodamine B (green light) [4], organic dyes [4] |
| Spectroscopic Probes | Detects reactive species generated from EDA complexes | Dihydroethidium (DHE, superoxide detection) [20], DCFH (ROS detection) [20] |
Advanced Applications in Synthesis
Workflow for Dual Bio-/Photo-Catalytic System
The following diagram illustrates the integrated workflow of a synergistic photo-/biocatalytic system where an EDA complex and an enzyme operate concurrently to achieve enantioselective transformations.
Key Applications and Performance Metrics
Recent research has demonstrated several innovative applications of EDA complexes in synthetic chemistry, particularly in challenging transformations.
Table 3: Advanced Synthetic Applications of EDA Complex Photochemistry
| Application | Reaction System | Key Performance Metrics | Significance |
|---|---|---|---|
| Aerobic Oxygenation | 1,2-Dinitrobenzene/tertiary amine EDA complex [20] | Converts boronic acids to phenols using O₂; 5 mol% acceptor loading; functional group tolerance | Overcomes traditional oxygen sensitivity of EDA complexes; enables superoxide generation |
| C–S Bond Formation | Thiophenol/aryl halide EDA complex [18] | Metal-free thioether synthesis; mild conditions; no catalyst required | Sustainable methodology for pharmacologically relevant C–S bonds |
| Enantioselective Hydroamination | ERED/Rhodamine B dual system [4] | Up to >99% ee; 82% yield; broad substrate scope | Demonstrates simultaneous control of chemo-, enantio-, and substrate selectivity |
| Triple Selectivity Control | Flavoprotein GkOYE-G7 [19] | >90% enantiomeric excess; predictable substrate scope | Reveals reaction-level rather than binding-based control mechanisms |
Radicals are highly reactive, short-lived chemical species that typically react indiscriminately with biological materials, posing a significant challenge for their use in selective synthesis. Despite this inherent reactivity, nature has evolved thousands of enzymes that employ radicals to catalyze thermodynamically challenging reactions essential to life. The fundamental challenge in harnessing these reactive intermediates lies in controlling their reactivity to achieve specific synthetic outcomes, particularly stereoselective transformations. Traditional chemical approaches often struggle with this control, leading to racemic mixtures or unwanted side reactions. Photoenzymatic catalysis has emerged as a powerful strategy to address this challenge, integrating the reactivity of photogenerated radicals with the exquisite stereocontrol of enzymatic environments to achieve unprecedented selectivity in asymmetric synthesis.
Fundamental Mechanisms of Enzymatic Radical Control
Electronic Control through SOMO-HOMO Inversion
Enzymes employ sophisticated quantum mechanical effects to modulate radical reactivity. Research on B12-dependent radical enzymes (glutamate mutase and methylmolonyl-Co-A mutase) has revealed a novel control mechanism involving non-Aufbau electronic structures. In these systems, the 5'-deoxyadenosyl (Ado•) radical exhibits SOMO-HOMO inversion (SHI), where the singly occupied molecular orbital (SOMO) is lower in energy than the highest occupied molecular orbital (HOMO) [21].
This electronic phenomenon is not merely a curiosity but serves a critical regulatory function. The magnitude of SHI is modulated through Coulombic interactions between the radical and a conserved glutamate residue (Glu-) within the active site. When the Ado• radical is first formed, hydrogen bonding with Glu- is minimal, resulting in more pronounced SHI and consequently lower reactivity. As the radical migrates toward the substrate (up to 8 Å), stronger hydrogen bonding with Glu- occurs, leading to less pronounced SHI and increased reactivity by 1-3 orders of magnitude [21]. This precise electronic tuning allows the enzyme to maintain control over the highly reactive radical intermediate throughout its catalytic journey.
Steric Control through Active Site Engineering
Beyond electronic effects, enzymes utilize precisely engineered binding pockets to enforce stereocontrol through spatial constraints. In photo-enzymatic cascade reactions for synthesizing α-functionalized phenylpyrrolidines, the binding pocket of engineered VHbCH carbene transferase provides a stable reaction environment that enhances enantioselectivity. Computational studies have confirmed that active pocket repositioning plays a critical role in stereoselective regulation, with the engineered enzyme achieving exceptional stereoselectivity (up to 99% ee) [22].
Similarly, in the asymmetric synthesis of hydroxysulfone compounds, engineering the substrate binding region of ketoreductase from Lactobacillus kefir (LkKRED) involved mutating residues within 5 Å of the substrate binding pocket. Residues S96, L153, and Y190 were identified as critical for stereocontrol, with the Y190S/S96T double variant achieving 99% yield and 99% ee by reducing steric hindrance and optimizing substrate orientation within the binding pocket [23].
Table 1: Performance of Engineered Enzymes in Stereoselective Radical Reactions
| Enzyme | Reaction Type | Modification | Yield (%) | Enantiomeric Excess (ee%) |
|---|---|---|---|---|
| Engineered SD-VHbCH carbene transferase | Photo-enzymatic C–H functionalization | Active pocket repositioning | N/R | 99% |
| LkKRED (Y190S/S96T variant) | Ketosulfone reduction | Semi-rational design of binding pocket | 99% | 99% |
| Wild-type LkKRED | Ketosulfone reduction | None | 7% | 71% |
N/R: Not reported in the sourced context
Experimental Protocols for Photo-Enzymatic Radical Reactions
Protocol 1: One-Pot Photo-Enzymatic Cascade for α-Functionalized Phenylpyrrolidine Synthesis
Principle: This protocol integrates a light-driven C–N cross-coupling reaction with biocatalytic carbene transfer to achieve enantioselective C(sp³)–H functionalization of saturated N-heterocyclic scaffolds [22].
Materials:
- Aryl bromides (various derivatives)
- Cyclic secondary amines
- Dual nickel/photoredox catalyst (Ni/PC)
- Engineered SD-VHbCH carbene transferase (whole-cell system)
- DMSO solvent
- Blue LED light source
- Inert atmosphere (argon or nitrogen)
Procedure:
- Photocatalytic Cross-Coupling:
- Charge a dried reaction vessel with aryl bromide (1.0 equiv), cyclic secondary amine (1.2 equiv), and Ni/PC catalyst (5 mol% each).
- Add anhydrous DMSO (0.1 M concentration relative to aryl bromide) under inert atmosphere.
- Irradiate with blue LEDs (450 nm) at room temperature for 12-24 hours with vigorous stirring.
- Monitor reaction completion by TLC or LC-MS.
Biocatalytic Carbene Transfer:
- Without intermediate purification, add the engineered SD-VHbCH whole-cell biocatalyst (20 mg/mL final concentration) directly to the reaction mixture.
- Add additional cofactors as required (specific composition may vary based on enzyme engineering).
- Incubate at 30°C with shaking at 200 rpm for 6-12 hours.
- Monitor enantioselectivity by chiral HPLC or SFC.
Workup and Purification:
- Extract the reaction mixture with ethyl acetate (3 × 50 mL).
- Combine organic layers, dry over anhydrous Na₂SO₄, and concentrate under reduced pressure.
- Purify the crude product by flash chromatography on silica gel.
- Analyze enantiomeric excess by chiral stationary phase HPLC.
Critical Notes:
- Maintain strict anaerobic conditions during the photocatalytic step to prevent catalyst deactivation.
- The one-pot nature avoids purification of reactive intermediates, minimizing decomposition pathways.
- Enzyme performance may vary with batch; preliminary activity assays are recommended.
Protocol 2: Photo-Biocatalytic Asymmetric Hydroamination
Principle: This protocol employs flavin-dependent ene-reductases to generate and control aminium radical cations for asymmetric intermolecular hydroamination, addressing a long-standing challenge in chemical synthesis [24].
Materials:
- Hydroxylamine precursor (alkyl-derived)
- Olefin substrate
- Flavin-dependent ene-reductase
- NAD(P)H cofactor (regeneration system optional)
- Phosphate buffer (pH 7.0)
- Visible light source (blue or green LEDs)
- Photoreactor with temperature control
Procedure:
- Reaction Setup:
- Prepare a solution of hydroxylamine precursor (1.0 equiv) and olefin substrate (1.5 equiv) in potassium phosphate buffer (50 mM, pH 7.0).
- Add purified ene-reductase or whole-cell biocatalyst (10 mg/mL final concentration).
- Include NADPH (0.5 mM) or NADH with regeneration system as required.
Photoirradiation:
- Transfer the reaction mixture to a photochemical reactor with temperature control.
- Irradiate with visible light (appropriate wavelength for enzyme photochemistry) at 25°C with stirring.
- Monitor reaction progress by TLC, GC, or LC-MS.
Enantioselective Hydroamination:
- The enzyme catalyzes both photogeneration of the aminium radical cation and enantioselective hydrogen atom transfer.
- Continue irradiation until complete consumption of starting material (typically 8-24 hours).
Product Isolation:
- Extract reaction mixture with ethyl acetate or dichloromethane (3 × volume).
- Dry combined organic layers over anhydrous MgSO₄.
- Concentrate under reduced pressure and purify by flash chromatography.
- Determine enantiomeric purity by chiral HPLC or GC.
Critical Notes:
- Enzyme selection is critical; different ene-reductases may yield opposite stereoselectivities.
- Light intensity should be optimized to balance radical generation with enzyme stability.
- Control experiments without light or enzyme confirm the photo-biocatalytic nature.
Visualization of Enzymatic Radical Control Mechanisms
SOMO-HOMO Inversion in Radical Enzyme Control
Diagram 1: Radical migration and reactivity modulation via SHI in B12 enzymes. SHI: SOMO-HOMO inversion [21].
Integrated Photo-Biocatalytic Cascade Workflow
Diagram 2: Integrated workflow for photo-biocatalytic radical taming [22] [23] [24].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Reagents for Photo-Enzymatic Radical Reactions
| Reagent/Material | Function/Role | Application Examples |
|---|---|---|
| Engineered Carbene Transferases (e.g., SD-VHbCH) | Stereoselective functionalization of C-H bonds | α-Functionalized phenylpyrrolidine synthesis [22] |
| Flavin-Dependent Ene-Reductases | Photogeneration and control of aminium radical cations | Asymmetric hydroamination [24] |
| Engineered Ketoreductases (e.g., LkKRED variants) | Stereoselective reduction of prochiral ketones | Hydroxysulfone synthesis [23] |
| Dual Nickel/Photoredox Catalyst (Ni/PC) | Light-driven C–N cross-coupling | In situ generation of N-heterocyclic scaffolds [22] |
| Ru(II) Photocages | Light-triggered enzyme inhibition with spatial control | Precise modulation of CYP enzyme activity [25] |
| NAD(P)H Cofactors | Redox equivalents for enzymatic catalysis | Ketoreductase-mediated asymmetric reductions [23] |
| Blue/Green LED Systems | Energy source for photochemical steps | Radical generation in photocatalytic steps [22] [23] |
The integration of photochemistry with enzymatic catalysis represents a paradigm shift in asymmetric synthesis, particularly for controlling highly reactive radical intermediates. Through sophisticated mechanisms like SOMO-HOMO inversion and precisely engineered active sites, enzymes can tame these fleeting species to achieve exceptional stereocontrol. The experimental protocols and tools outlined herein provide researchers with practical approaches to implement these strategies for synthesizing valuable chiral building blocks. As protein engineering capabilities advance and our understanding of radical enzymology deepens, this synergistic approach will undoubtedly expand to encompass an even broader range of transformations, further solidifying its role in sustainable pharmaceutical synthesis and green manufacturing.
Synthetic Applications: Building Complex Chiral Molecules
Synthesis of Fluorinated Amides with Remote Stereocenters
The incorporation of fluorine atoms into organic molecules, particularly amides, is a powerful strategy in modern medicinal chemistry and drug development. Fluorinated amides serve as prevalent structural motifs in pharmaceuticals and bioactive molecules, with over 50% of modern pharmaceuticals containing fluorine by 2023 [5]. The introduction of fluorine can significantly alter a compound's physical properties, metabolic stability, and biological activity due to fluorine's high electronegativity and favorable biocompatibility [5]. However, the enantioselective synthesis of these compounds, especially those bearing challenging remote stereocenters, remains a significant challenge for synthetic chemists [5] [26].
This Application Note focuses specifically on photoenzymatic strategies for synthesizing fluorinated amides with remote stereocenters, highlighting recent breakthroughs in enzyme catalysis that overcome previous limitations in stereocontrol. These methodologies provide sustainable and efficient approaches to access valuable fluorinated building blocks for drug development programs.
Photoenzymatic Enantioselective Synthesis
Ene-Reductase Catalyzed Hydroalkylation
A groundbreaking visible-light-driven ene-reductase system has been developed for the enantioselective synthesis of α-fluorinated amides with distal chirality. This system utilizes flavin-dependent 'ene'-reductases (ERs) to generate carbon-centered radicals from fluorine-containing brominated amides under blue light irradiation, enabling their enantioselective hydroalkylation with alkenes [5].
Key Reaction Components:
- Catalyst: Ene-reductases (OYE1, OYE2, OYE3, XenA, XenB)
- Cofactor System: Flavin mononucleotide (FMN), glucose dehydrogenase (GDH), D-(+)-glucose, NADP+
- Light Source: Blue LED (455-465 nm)
- Radical Precursor: Fluorine-containing brominated amides (e.g., F-1)
- Coupling Partners: Electron-deficient alkenes
Table 1: Optimization of Reaction Parameters for Photoenzymatic Hydroalkylation
| Parameter | Initial Condition | Optimized Condition | Impact on Yield/ee |
|---|---|---|---|
| Light Source | 12 W, 455 nm | 45 W, 465 nm | Yield increased from 15% to 21% |
| Buffer System | Sodium phosphate (NaPi) | Tris-HCl (pH 8.5) | Yield increased ~1.7 times |
| Enzyme Loading | Baseline | 2 mol% OYE1 | Yield increased to 51% |
| Enzyme Variant | Wild-type OYE1 | Y375F mutant | Yield >90% with maintained ee >90% |
The reaction optimization demonstrated that alkaline conditions (Tris-HCl, pH 8.5) and increased enzyme loading significantly improved reaction efficiency. Through systematic enzyme engineering, the Y375F mutation was identified as crucial for enhancing yield while maintaining excellent enantioselectivity [5].
Experimental Protocol
Procedure for Photoenzymatic Hydroalkylation of Fluorinated Amides:
Reaction Setup: In a 5 mL reaction vial, add the following components:
- Fluorinated bromoamide substrate F-1 (0.1 mmol, 1.0 equiv)
- Alkene coupling partner (0.12 mmol, 1.2 equiv)
- Selected ene-reductase (2 mol%)
- FMN (0.5 mol%)
- NADP+ (0.5 mol%)
- GDH (2 mg)
- D-(+)-glucose (5.0 equiv)
- Tris-HCl buffer (2.0 mL, 50 mM, pH 8.5)
Photoreaction: Seal the vial and irradiate with blue LEDs (45 W, 465 nm) with continuous stirring for 24-48 hours at 25°C.
Reaction Monitoring: Monitor reaction progress by TLC or GC-MS until complete consumption of the starting material is observed.
Workup: Extract the reaction mixture with ethyl acetate (3 × 5 mL), combine organic layers, dry over anhydrous Na₂SO₄, and concentrate under reduced pressure.
Purification: Purify the crude product by flash column chromatography on silica gel to obtain the desired fluorinated amide with remote stereocenter.
Analysis: Determine enantiomeric excess by chiral HPLC or SFC analysis.
This protocol has been successfully applied to synthesize diversified α-fluorinated amides with high yields (up to 91%) and excellent distal chirality (up to 97% enantiomeric excess) [5].
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Photoenzymatic Fluorinated Amide Synthesis
| Reagent/Enzyme | Function | Application Notes |
|---|---|---|
| Ene-reductases (OYE1) | Catalyzes enantioselective radical addition | Y375F mutant shows enhanced activity |
| Flavin Mononucleotide (FMN) | Photosensitizer and redox cofactor | Enables radical generation under light |
| Glucose Dehydrogenase (GDH) | Cofactor regeneration system | Maintains NADPH levels for sustained catalysis |
| α-Fluorinated Bromoamides | Radical precursors | Amide group crucial for enzyme binding and reactivity |
| Tris-HCl Buffer (pH 8.5) | Reaction medium | Alkaline conditions enhance reaction efficiency |
| Blue LED Light Source | Radical initiation | 45W, 465nm optimal for excitation |
Reaction Mechanism and Workflow
The photoenzymatic mechanism involves a sophisticated interplay between light absorption, radical generation, and enantioselective bond formation within the enzyme active site.
Diagram 1: Photoenzymatic Reaction Mechanism
The catalytic cycle begins with blue light absorption by the flavin cofactor, generating an excited state that initiates single electron transfer to the fluorinated bromoamide substrate. This process generates a carbon-centered radical that adds to the alkene coupling partner. The resulting radical intermediate undergoes enantioselective hydrogen atom transfer (HAT) within the enzyme active site, where precisely positioned amino acid residues control the stereochemical outcome at the remote center [5].
Diagram 2: Experimental Workflow
Comparative Analysis and Applications
The photoenzymatic approach represents a significant advancement over traditional methods for synthesizing fluorinated amides with remote stereocenters. Conventional strategies often relied on stoichiometric chiral auxiliaries or faced limitations in controlling remote stereochemistry [27]. The enzyme-based system provides exceptional control over stereoselectivity while operating under mild, environmentally friendly conditions.
Key Advantages:
- High Enantioselectivity: Up to 97% ee for γ-stereocenters relative to fluorine
- Broad Substrate Scope: Accommodates various fluorinated amides and alkene coupling partners
- Sustainability: Utilizes visible light as energy source and operates at ambient temperature
- Tunability: Enzyme engineering enables optimization for specific substrates
This methodology has significant implications for drug discovery, particularly in accessing chiral fluorinated 3D molecules that exhibit enhanced biological activity, metabolic stability, and membrane permeability – critical properties for pharmaceutical development [26]. The ability to precisely install fluorine atoms at internal positions with controlled stereochemistry enables medicinal chemists to fine-tune molecular properties while maintaining three-dimensional complexity.
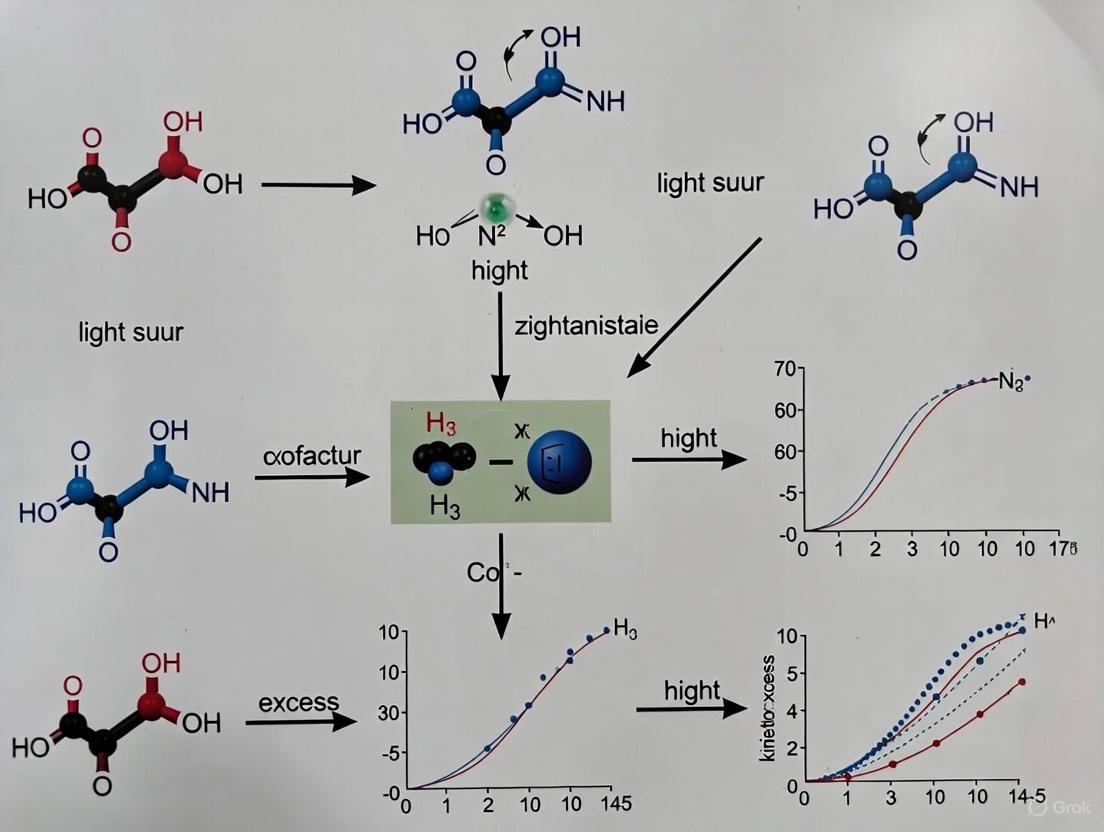
Intermolecular Radical Hydroamination for Chiral Amine Synthesis
The synthesis of chiral amines, which are pivotal structural motifs in pharmaceuticals, agrochemicals, and natural products, represents a central challenge in modern organic chemistry. Intermolecular radical hydroamination offers a powerful, atom-economical route to construct these valuable scaffolds directly from simple alkenes and amine precursors. However, controlling the reactivity of transient radical intermediates and achieving high levels of enantioselectivity has been a long-standing obstacle. Traditional catalytic systems struggle to suppress deleterious side reactions, such as α-deprotonation and hydrogen atom abstraction, which compete with the desired pathway [28].
Photoenzymatic catalysis has emerged as a transformative platform to address these challenges. By repurposing natural enzymes, particularly flavin-dependent 'ene'-reductases (EREDs), chemists can now generate and harness highly reactive nitrogen-centered radicals under mild conditions. This approach leverages the enzyme's innate chiral environment to enforce enantioselectivity, enabling asymmetric intermolecular hydroamination reactions that were previously inaccessible. This Application Note details the experimental protocols and mechanistic insights for implementing this cutting-edge methodology, contextualized within the broader thesis of advancing photoenzymatic enantioselective synthesis.
Core Principles and Mechanistic Insights
The Catalytic Cycle
The photoenzymatic hydroamination mechanism involves a carefully orchestrated sequence of steps within the enzyme's active site, centered on the flavin mononucleotide (FMN) cofactor.
The mechanism initiates with photoexcitation of the reduced flavin mononucleotide (FMNH⁻) to FMNH⁻*, a potent reductant. This excited species transfers an electron to a nitrogen-containing precursor, typically featuring a labile N–O bond (e.g., a hydroxylamine derivative with a phenolate leaving group). This transfer triggers homolytic cleavage to generate an aminium radical cation (ARC). This electrophilic radical adds across the alkene substrate, forming a new C–N bond and a carbon-centered radical. Finally, the enzyme mediates an enantioselective hydrogen atom transfer (HAT) from the flavin semiquinone (FMNH•) to this nascent radical, furnishing the enantioenriched amine product and regenerating the oxidized FMN cofactor, which is subsequently reduced by the NADPH/glucose dehydrogenase (GDH) regeneration system [3].
Advantages of the Photoenzymatic System
- > Unprecedented Enantiocontrol: The confined chiral pocket of the ene-reductase exerts exquisite control over the hydrogen atom transfer step, leading to high enantiomeric excess (ee), often exceeding 90% and reaching up to 99% in optimized systems [3] [22].
- > External Photocatalyst-Free: Unlike many radical hydroamination methods, the flavin cofactor acts as the internal photocatalyst, simplifying the reaction mixture and purifying the product [3].
- > Mild Reaction Conditions: Reactions proceed efficiently at room temperature under visible light irradiation in aqueous or mixed aqueous-organic buffers, compatible with a range of functional groups [5] [3].
Key Research Reagent Solutions
The following table catalogues the essential reagents and materials critical for setting up a photoenzymatic hydroamination reaction.
Table 1: Essential Research Reagents for Photoenzymatic Hydroamination
| Reagent/Material | Function/Role | Key Details & Examples |
|---|---|---|
| Ene-Reductase (ERED) | Biocatalyst; Provides chiral environment & flavin cofactor. | XenB from Pseudomonas putida; OYE1; Engineered variants (e.g., XenB-W100L, XenB-A232F) for improved performance [3]. |
| Flavin Cofactor (FMN) | Internal photosensitizer & redox mediator. | Flavin Mononucleotide (FMN); recycled in situ by NADPH [3]. |
| Cofactor Regeneration System | Sustains catalytic cycle by reducing FMN. | NADP⁺/Glucose Dehydrogenase (GDH)/D-Glucose; maintains supply of FMNH⁻ [5] [3]. |
| Nitrogen Radical Precursor | Source of the aminium radical cation (ARC). | Hydroxylamines with phenolate leaving groups (e.g., 4-cyanophenolate). Carbamates for protected primary amines [3]. |
| Alkene Substrate | Radical acceptor for C–N bond formation. | Electron-rich alkenes (e.g., ɑ-methyl styrene derivatives); regioselectivity is anti-Markovnikov due to electrophilic ARC [3] [29]. |
| Reaction Buffer | Aqueous reaction medium with controlled pH. | Tris-HCl (pH ~8.5) or Imidazole (pH ~6.5); optimal pH is enzyme and substrate-dependent [5] [3]. |
Quantitative Performance Data
The performance of photoenzymatic hydroamination has been quantitatively demonstrated across a broad range of substrates. The data below summarizes key metrics for yield and enantioselectivity.
Table 2: Performance Summary of Photoenzymatic Hydroamination [28] [5] [3]
| Substrate Class | Example Structure | Representative Yield (%) | Representative ee (%) | Optimal Enzyme |
|---|---|---|---|---|
| Protected Aliphatic Amines | N-Carbamyl pyrrolidines & piperidines | 25 – 88 | 93 – 97 | XenB-A232L (E2) [3] |
| Aromatic Alkenes | para-Substituted styrenes | 50 – 96 | 88 – 97 | XenB-A232L (E2) [3] |
| Fluorinated Amides | α-Difluoroamides | Up to 91 | Up to 97 | OYE1-Y375F [5] |
| Heteroaryl Amines | Aminopyrimidines & Aminopyridines | 57 – 89* | N/A* | [Ir-B]OTf (Chemical PC) [29] |
| Sulfonamides | Aryl & Alkyl Sulfonamides | Up to 97* | N/A* | [Ir-A]OTf/[Ir-B]OTf (Chemical PC) [30] |
Note: Reactions for Heteroaryl Amines and Sulfonamides typically employ synthetic iridium photocatalysts ([Ir-A]OTf, [Ir-B]OTf) and proceed with excellent anti-Markovnikov selectivity, but enantioselectivity is not reported for the racemic versions shown here. Yields are high, demonstrating the robustness of the radical mechanism [29] [30].
Detailed Experimental Protocol
Photoenzymatic Hydroamination Using Evolved Ene-Reductase
This protocol describes the intermolecular hydroamination between carbamate 1a and ɑ-methyl styrene using an evolved XenB variant (E2, XenB-A232L) to produce chiral amine 3a in high yield and enantioselectivity [3].
Materials:
- Enzyme: Purified XenB-A232L mutant (E2) lyophilized powder [3].
- Cofactors: NADP⁺ (disodium salt), FMN (disodium salt).
- Regeneration System: Glucose Dehydrogenase (GDH, from Pseudomonas sp.), D-(+)-Glucose.
- Substrates: Hydroxylamine precursor 1a (e.g., with 4-cyanophenolate leaving group), ɑ-methyl styrene.
- Solvents & Buffers: Imidazole buffer (1.0 M, pH 6.5), deionized water.
Procedure:
- Reaction Setup: In a 4 mL clear glass vial equipped with a magnetic stir bar, combine the following:
- Imidazole Buffer (1.0 M, pH 6.5): 200 µL (Final conc. 100 mM)
- NADP⁺ (10 mM stock in H₂O): 50 µL (Final 0.1 mM)
- FMN (5 mM stock in H₂O): 20 µL (Final 0.02 mM)
- D-Glucose (1.0 M stock in H₂O): 50 µL (Final 100 mM)
- GDH (5 mg/mL stock in buffer): 20 µL (∼2.5 U)
- Substrate 1a (0.1 M stock in DMSO): 50 µL (0.005 mmol, 1.0 equiv)
- ɑ-Methyl styrene (0.1 M stock in DMSO): 150 µL (0.015 mmol, 3.0 equiv)
- Deionized H₂O: 450 µL
- Enzyme XenB-A232L (E2, from 10 µM stock in buffer): 50 µL (2 mol%)
Initiation and Irradiation:
- Cap the vial and place it on a magnetic stirrer.
- Irradiate the reaction mixture with a blue LED strip (∼465 nm, 45 W) positioned approximately 5 cm from the vial.
- Stir the reaction vigorously at room temperature (25 °C) for 16-24 hours.
Work-up:
- Quench the reaction by adding saturated aqueous NaCl solution (1 mL).
- Extract the aqueous mixture with ethyl acetate (3 × 2 mL).
- Combine the organic extracts and dry over anhydrous MgSO₄.
- Filter and concentrate the organic layer under reduced pressure.
Purification and Analysis:
- Purify the crude residue by flash column chromatography on silica gel.
- Analyze the enantiomeric purity of the product 3a by chiral HPLC or GC.
- Expected Outcome: The product 3a can be obtained in 88% yield and 93% ee [3].
Application Notes for Drug Development
- Handling Sensitive Intermediates: Nitrogen radical precursors, especially those with N–O bonds, can be sensitive to heat and light. Store stock solutions in the dark at -20 °C and prepare them fresh weekly. Reactions should be set up in low-actinic glassware if available.
- Optimization and Scale-up: For new substrate combinations, a brief optimization of pH (testing buffers from pH 6.0-9.0) and enzyme loading (1-5 mol%) is recommended. The reaction has been successfully demonstrated on gram-scale, showing robust performance for potential industrial application [3].
- Analytical Techniques: Monitor reaction progress by TLC or LC-MS. Determine enantiomeric excess (ee) using Chiral Stationary Phase HPLC or GC. Absolute configuration can be assigned by comparing optical rotation with literature values or via X-ray crystallography of derivative compounds.
Biosynthesis of Chiral Vicinal Diamines via Dual Catalysis
Chiral vicinal diamines are fundamental structural motifs found in numerous natural products, pharmaceuticals, and chiral ligands or auxiliaries for asymmetric catalysis [4]. Despite their importance, the catalytic enantioselective synthesis of these molecules, particularly via direct hydroamination of enamines, remains a significant challenge [4]. Traditional chemical methods often rely on transition metal complexes or chiral phosphoric acids via two-electron electrophilic pathways [4]. In recent years, photoenzymatic catalysis has emerged as a powerful strategy to address enantioselectivity challenges associated with photoinduced radical reactions, combining the remarkable stereocontrol of enzymes with the versatile reactivity of photocatalysts [4] [31]. This application note details a novel dual bio-/photo-catalytic system for achieving enantioselective hydroamination of enamines, providing researchers with robust protocols for synthesizing valuable chiral vicinal diamines with exceptional stereochemical control.
Experimental Protocols
Dual Photo-/Bio-Catalytic System for Enantioselective Hydroamination
This protocol describes the synergistic use of an ene-reductase (ERED) and an organic photocatalyst for the enantioselective synthesis of vicinal diamines via nitrogen-centered radical (NCR) hydroamination [4].
Reagents and Materials
- Enzyme: GluER-T36A-Y177F-F269V-K277M-Y343F (ER-M5) mutant [4]
- Photocatalyst: Rhodamine B (RhB) [4]
- NCR Precursor: N-amidopyridinium salt (e.g., 2a) [4]
- Radical Acceptor: N-(1-phenylvinyl)acetamide (e.g., 1a) [4]
- Cofactor Regeneration System: Glucose dehydrogenase (GDH), NADP+, D-glucose [4]
- Solvent: Phosphate buffer (50 mM, pH 7.5) or other appropriate aqueous buffer [4]
- Light Source: Green LEDs (530-540 nm) [4]
Procedure
Reaction Setup: In a 4 mL glass vial equipped with a magnetic stir bar, combine the following components sequentially:
- Phosphate buffer (50 mM, pH 7.5, final volume 1 mL)
- N-(1-phenylvinyl)acetamide 1a (0.05 mmol, 1.0 equiv)
- N-amidopyridinium salt 2a (0.075 mmol, 1.5 equiv)
- Rhodamine B (2 mol%)
- ER-M5 enzyme (1 mol%)
- GDH (5 mg/mL), NADP+ (0.5 mM), and D-glucose (10 mM)
Reaction Conditions:
- Seal the vial with a septum and place it 5 cm from a green LED array (530-540 nm)
- Stir the reaction mixture at 25°C for 24-48 hours
- Monitor reaction progress by TLC, HPLC, or LC-MS
Workup and Purification:
- After completion, extract the reaction mixture with ethyl acetate (3 × 5 mL)
- Combine the organic layers and dry over anhydrous Na₂SO₄
- Concentrate under reduced pressure
- Purify the crude product by flash column chromatography (silica gel, hexane/ethyl acetate) to afford the desired vicinal diamine product (S)-3a
Analysis
- Yield: 72% isolated yield [4]
- Enantiomeric Excess: 98% ee (determined by chiral HPLC) [4]
- Absolute Configuration: (S) (assigned by single-crystal X-ray diffraction) [4]
Key Optimization Strategies
The development of this protocol involved critical optimization of several parameters to achieve high yield and enantioselectivity:
- Enzyme Engineering: Site-directed mutagenesis of native GluER created the quintuple mutant ER-M5 (T36A-Y177F-F269V-K277M-Y343F) with enhanced activity and selectivity [4]
- Photocatalyst Screening: Rhodamine B was identified as the optimal photocatalyst for generating NCRs under green light excitation [4]
- Wavelength Optimization: Green light (530-540 nm) provided better compatibility with the biological system and reduced side reactions compared to blue or UV light [4]
- Cofactor Regeneration: The GDH/glucose/NADP+ system maintained efficient flavin reduction throughout the reaction [4]
Data Presentation
Substrate Scope and Performance
Table 1: Substrate scope for the enantioselective hydroamination of enamides with N-amidopyridinium salts under optimized dual catalytic conditions [4]
| Product | R₁ Substituent | R₂ Substituent | Yield (%) | ee (%) |
|---|---|---|---|---|
| 3a | H | 4-OMe-C₆H₄ | 72 | 98 |
| 3b | 2-OMe | 4-OMe-C₆H₄ | 78 | >99 |
| 3c | 3-OMe | 4-OMe-C₆H₄ | 82 | 98 |
| 3d | 4-F | 4-OMe-C₆H₄ | 65 | 97 |
| 3e | 4-Cl | 4-OMe-C₆H₄ | 68 | 97 |
| 3f | 4-Br | 4-OMe-C₆H₄ | 63 | 98 |
| 3g | 4-CF₃ | 4-OMe-C₆H₄ | 58 | 96 |
| 3h | 2-Naphthyl | 4-OMe-C₆H₄ | 45 | 90 |
| 3i | H | 4-Cl-C₆H₄ | 70 | 98 |
| 3j | H | 4-CN-C₆H₄ | 65 | 97 |
| 3k | H | 2-Thienyl | 62 | 96 |
Table 2: Key research reagent solutions for photoenzymatic diamine synthesis
| Reagent | Function | Application Notes |
|---|---|---|
| ERED Enzymes (GluER mutants, OYE1) [4] [5] | Enantioselective radical hydroamination and hydrogen atom transfer | Site-directed mutagenesis enhances activity and selectivity; FMN cofactor required |
| Organic Photocatalysts (Rhodamine B) [4] | Green light absorption and NCR generation from N-amidopyridinium salts | Enables radical initiation under biologically compatible conditions |
| N-Amidopyridinium Salts [4] | Nitrogen-centered radical (NCR) precursors | N-N bond cleavage under photochemical conditions generates amidyl radicals |
| Flavin Cofactors (FMN) [5] [4] | Natural photoenzyme cofactor | Participates in single-electron transfer processes and hydrogen atom transfer |
| Cofactor Regeneration System (GDH/Glucose/NADP+) [4] [5] | Sustained enzymatic activity by maintaining reduced flavin state | Essential for prolonged reaction efficiency and conversion |
Visualization of Workflows and Mechanisms
Dual Catalytic Cycle Mechanism
Dual Catalytic Cycle: This diagram illustrates the synergistic mechanism between the photocatalyst (RhB) and the ene-reductase (ERED). The photocatalyst generates nitrogen-centered radicals under green light, which add to enamides. The ERED then controls the enantioselective hydrogen atom transfer to form the chiral diamine product, with the flavin cofactor (FMN) being continuously regenerated by the enzymatic reduction system [4].
Experimental Workflow for Photoenzymatic Diamine Synthesis
Experimental Workflow: This flowchart outlines the key steps in the photoenzymatic synthesis of chiral vicinal diamines, from reaction setup through irradiation, radical generation, enantioselective bond formation, and final product isolation and analysis. The dashed line indicates the simultaneous cofactor regeneration essential for maintaining enzymatic activity [4].
Discussion
The dual photo-/bio-catalytic system represents a significant advancement in asymmetric synthesis, addressing longstanding challenges in controlling highly reactive nitrogen-centered radicals for enantioselective bond formation [4]. The combination of ene-reductases with organic photocatalysts creates a synergistic platform that merges the exceptional stereocontrol of enzymes with the versatile radical generation capability of photoredox catalysis.
Key advantages of this methodology include:
- Exceptional Enantiocontrol: The enzyme active pocket provides a highly chiral environment for radical taming, achieving >99% ee in many cases [4]
- Mild Reaction Conditions: Green light irradiation and ambient temperature minimize substrate decomposition and enzyme denaturation [4]
- Broad Functional Group Tolerance: The system accommodates various electron-donating and electron-withdrawing substituents on both reaction partners [4]
- Scalability: The protocol has been demonstrated on 0.1 mmol scale without erosion of enantioselectivity [4]
Mechanistic studies confirmed the radical nature of this transformation through radical trapping experiments and characterization of the electron donor-acceptor complex between the reduced enzyme and the N-amidopyridinium salt [4]. The unique ability of flavin-dependent ene-reductases to control the stereochemistry of radical intermediates underscores their potential beyond natural enone reduction activities.
This protocol provides pharmaceutical and process chemists with a robust method for synthesizing enantiopure vicinal diamines, valuable building blocks in drug discovery and development. The principles established here may guide future developments in photoenzymatic catalysis for other challenging asymmetric transformations.
Multi-Component Cascade Reactions for Functionalized Heterocycles
The synthesis of functionalized heterocycles represents a core pursuit in modern pharmaceutical research, as these structures are ubiquitous in bioactive molecules and therapeutic agents. Multi-component cascade reactions have emerged as a powerful strategy to construct these complex scaffolds efficiently, allowing for a rapid increase in molecular complexity from simple starting materials in a single operation. The integration of photocatalysis with enzyme catalysis has been particularly transformative, enabling access to radical intermediates under mild conditions while maintaining exquisite stereocontrol. This application note details cutting-edge protocols within the broader context of photoenzymatic enantioselective synthesis, providing researchers with practical methodologies for synthesizing valuable chiral heterocycles. These approaches address long-standing challenges in sustainable synthesis, including the need for toxic reagents, precious metal catalysts, and lengthy synthetic sequences, thereby offering more direct routes to pharmaceutically relevant scaffolds.
Experimental Protocols & Data
This section provides detailed experimental methodologies for three distinct photoenzymatic multi-component reactions, enabling the synthesis of fluorinated amides, vicinal diamines, and functionalized pyrrolidines.
Photoenzymatic Synthesis of Fluorinated Amides
This protocol describes an enantioselective hydroalkylation for synthesizing α-fluorinated amides with distal chirality, using a visible-light-driven ene-reductase system [5].
Reaction Setup: In a 4 mL glass vial, combine the following reagents:
- Fluorine-containing brominated amide substrate (F-1, 0.1 mmol, 1.0 equiv)
- Electron-rich alkene (0.12 mmol, 1.2 equiv)
- OYE1 enzyme variant (Y375F mutant, 2 mol%)
- Flavin Mononucleotide (FMN, 2 mol%)
- NADP⁺ (0.5 mg)
- Glucose Dehydrogenase (GDH, 2 mg)
- D-(+)-Glucose (0.02 mmol)
- Tris-HCl Buffer (2 mL, pH 8.5)
Reaction Procedure:
- Seal the vial with a septum cap.
- Purge the reaction mixture with nitrogen gas for 5 minutes.
- Irradiate the mixture with 45 W blue LEDs (λ = 465 nm) while stirring at 25°C for 24 hours.
- Monitor reaction progress by TLC or GC-MS.
- Upon completion, extract the aqueous layer with ethyl acetate (3 × 5 mL).
- Combine the organic extracts, dry over anhydrous MgSO₄, filter, and concentrate under reduced pressure.
- Purify the crude product by flash column chromatography on silica gel.
Key Analytical Data: The protocol yields diversified α-fluorinated amides in up to 91% yield and 97% enantiomeric excess (ee). Stereoselectivity is determined by chiral HPLC or GC analysis [5].
Dual Photo/Biocatalytic Synthesis of Vicinal Diamines
This method enables the enantioselective hydroamination of enamides using a synergistic catalytic system comprising an ene-reductase and an organic dye, providing access to valuable chiral vicinal diamines [4].
Reaction Setup: In a 4 mL glass vial, combine:
- N-(1-phenylvinyl)-acetamide derivative (1a, 0.1 mmol, 1.0 equiv)
- N-amidopyridinium salt (2a, 0.12 mmol, 1.2 equiv)
- Engineered ene-reductase (GluER-M5 variant, 1 mol%)
- Rhodamine B (RhB, 2 mol%)
- NADP⁺ (0.5 mg)
- Glucose Dehydrogenase (GDH, 2 mg)
- D-Glucose (0.02 mmol)
- Phosphate Buffer (2 mL, pH 7.5)
Reaction Procedure:
- Seal and purge the vial with nitrogen.
- Irradiate with 530-540 nm green LEDs while stirring at 25°C for 20 hours.
- Quench the reaction by adding saturated aqueous NH₄Cl (5 mL).
- Extract with ethyl acetate (3 × 5 mL).
- Dry the combined organic layers over Na₂SO₄, filter, and concentrate.
- Purify the residue via flash chromatography.
Key Analytical Data: This transformation proceeds with excellent enantioselectivity (up to >99% ee) and provides moderate to good yields (up to 82%). The absolute configuration of the diamines is assigned as (S) by single-crystal X-ray diffraction [4].
One-Pot Photo-enzymatic Cascade for α-Functionalized Phenylpyrrolidine
This one-pot protocol integrates a light-driven C–N cross-coupling with biocatalytic carbene transfer for the enantioselective C(sp³)–H functionalization of saturated N-heterocycles [22].
Reaction Setup - Step 1 (Photocatalytic Cross-Coupling):
- In a Schlenk tube, combine:
- Aryl bromide (0.2 mmol, 1.0 equiv)
- Cyclic secondary amine (e.g., pyrrolidine, 0.24 mmol, 1.2 equiv)
- Dual Ni/Photocatalyst system (5 mol%)
- DMSO (2 mL)
- Irradiate with blue LEDs under nitrogen atmosphere at room temperature for 12 hours to generate saturated N-heterocyclic intermediate in situ.
- In a Schlenk tube, combine:
Reaction Setup - Step 2 (Biocatalytic Functionalization):
- To the same pot, add:
- Whole-cell system containing engineered SD-VHbCH carbene transferase
- Ethyl diazoacetate (0.3 mmol, 1.5 equiv)
- Incubate the mixture at 30°C with shaking for 24 hours.
- To the same pot, add:
Workup and Isolation:
- Centrifuge the reaction mixture to remove cells.
- Extract the aqueous supernatant with ethyl acetate (3 × 10 mL).
- Dry the combined organic extracts, concentrate, and purify by flash chromatography.
Key Analytical Data: This cascade process affords α-functionalized phenylpyrrolidines with superior stereoselectivity (up to 99% ee) [22].
Table 1: Summary of Photoenzymatic Multi-Component Reactions for Heterocycle Synthesis
| Reaction Type | Catalytic System | Key Heterocycle Formed | Reported Yield | Reported ee | Light Source |
|---|---|---|---|---|---|
| Hydroalkylation | Ene-reductase (OYE1-Y375F) | α-Fluorinated Amides [5] | Up to 91% | Up to 97% | 45 W Blue LEDs (465 nm) |
| Hydroamination | Ene-reductase (GluER-M5) / Rhodamine B | Vicinal Diamines [4] | Up to 82% | Up to >99% | Green LEDs (530-540 nm) |
| Carbene Functionalization | Ni/PC & Carbene Transferase | α-Functionalized Phenylpyrrolidine [22] | Not Specified | Up to 99% | Blue LEDs |
Table 2: Key Reagent Solutions for Photoenzymatic Cascades
| Reagent / Cofactor | Function in the Reaction | Typical Loading |
|---|---|---|
| Flavin Mononucleotide (FMN) | Natural photoenzyme cofactor; mediates single-electron transfer and hydrogen atom transfer [5] [4] | 2 mol% |
| NADP⁺ / NADPH | Biological redox cofactor; provides reducing equivalents for enzyme turnover | 0.5 mg / in situ regeneration |
| Glucose Dehydrogenase (GDH) | Cofactor regeneration system; recycles NADP⁺ to NADPH using glucose as a sacrificial electron donor [5] [4] | 2 mg |
| D-(+)-Glucose | Sacrificial electron donor for in situ NADPH regeneration [5] [4] | 0.02 mmol |
| Rhodamine B (RhB) | Organophotoredox catalyst; generates N-centered radicals under green light [4] | 2 mol% |
Experimental Workflow and Signaling Pathway
The following diagram illustrates the general catalytic cycle and experimental workflow for a synergistic photo/biocatalytic system, as exemplified by the synthesis of vicinal diamines [4].
The mechanism involves a synergistic cycle where an organic photocatalyst (e.g., Rhodamine B) and an ene-reductase (ERED) operate in concert [4]. The photocatalyst, excited by green light, generates a nitrogen-centered radical (NCR) from a precursor like an N-amidopyridinium salt. This NCR adds to an enamide substrate. The resulting carbon-centered radical then enters the enzyme's active site, where the reduced flavin hydroquinone (FMNhq) donates a hydrogen atom via an enantiodetermining Hydrogen Atom Transfer (HAT) step. This key step, controlled by the engineered enzyme pocket, yields the chiral product and returns the flavin to its oxidized state, ready for reduction by the NADPH regeneration system [5] [4].
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions and Materials
| Category | Item | Specific Example / Notes | Critical Function |
|---|---|---|---|
| Enzymes | Ene-reductases (EREDs) | OYE1, GluER, XenA [5] [4] | Catalyze enantioselective H-atom transfer; engineered for activity/selectivity. |
| Carbene Transferases | SD-VHbCH variant [22] | Catalyze enantioselective carbene insertion into C-H bonds. | |
| Cofactors & Regeneration | Flavin Mononucleotide (FMN) | 2 mol% loading [5] | Photosensitizer and radical mediator within the enzyme. |
| NADP⁺ / NADPH | In situ regeneration preferred [5] [4] | Essential biological redox cofactor for enzyme reduction. | |
| Glucose Dehydrogenase (GDH) & D-Glucose | Standard regeneration system [5] | Maintains catalytic NADPH levels cost-effectively. | |
| Photocatalysts | Organophotoredox Catalyst | Rhodamine B (for green light) [4] | Generates radicals under mild, biocompatible light. |
| Transition Metal Photocatalyst | fac-Ir(ppy)₃ analogues (for blue light) [5] | Often used in initial photocatalytic steps. | |
| Radical Precursors | Halogenated Compounds | Bromodifluoroacetamides (e.g., F-1) [5] | Source of fluorinated carbon-centered radicals. |
| N-Centered Radical Precursors | N-Amidopyridinium salts [4] | Source of reactive NCRs for amination. | |
| Reaction Setup | LED Light Source | Blue (465 nm), Green (530 nm) [5] [4] | Provides specific wavelength for photoexcitation. |
| Buffer Systems | Tris-HCl (pH 8.5), Phosphate (pH 7.5) [5] [4] | Maintains optimal pH for enzyme activity and stability. |
Photoenzymatic catalysis has established itself as a powerful strategy for asymmetric synthesis, merging the energy of light with the exceptional stereocontrol of enzymes to access valuable chiral building blocks. A critical frontier in this field is the expansion of substrate scope, particularly the identification of tolerated functional groups and olefin types, which directly dictates the synthetic utility and applicability of these biocatalytic systems. This application note consolidates recent experimental data and detailed protocols to guide researchers in leveraging photoenzymatic platforms for the enantioselective synthesis of fluorinated amides and hydroarylation products, highlighting the specific functional groups and olefins compatible with these advanced methodologies.
Summarized Quantitative Data on Substrate Scope
The following tables summarize quantitative data on the tolerance of various functional groups and olefins in two distinct photoenzymatic systems: the synthesis of fluorinated amides using an ene-reductase system and the hydroarylation of olefins using an engineered Baeyer-Villiger Monooxygenase.
Table 1: Substrate Scope in Photoenzymatic Synthesis of Fluorinated Amides with Ene-Reductases [5]
| Substrate Class / Functional Group | Example Substituent (R) | Yield (%) | Enantiomeric Excess (ee %) |
|---|---|---|---|
| Electron-Rich Arenes | Not Specified | Up to 91 | Up to 97 |
| Bromodifluoroacetamide Derivative | F-1 (Precursor) | Successful Radical Generation | N/A |
| Ester Control Substrate | Ethyl Bromodifluoroacetate (F-0) | No Product Formation | N/A |
Table 2: Substrate Scope in Stereoselective Photoenzymatic Hydroarylation with Engineered BVMO (AcHYAR) [32]
| Substrate Region | Specific Functional Group / Substituent | Yield (%) | Enantiomeric Ratio (e.r.) |
|---|---|---|---|
| Aniline Motif (Electron-Rich) | 4-OMe, 5-OMe, 6-OMe, 7-OMe | High Yields | Good to Excellent |
| Aniline Motif (Bulky) | 6-NHBoc | 35 | 59:41 |
| Aniline Motif (Heteroaromatic) | Pyridine | Compatible (Yield affected by isomer) | Not Specified |
| Aniline Motif (Meta-Substituted) | Methyl | Good Yield | Low Regioselectivity (2.3:1) |
| Methoxy | Good Yield | Low Regioselectivity (1:1) | |
| Isopropyl | Good Yield | High Regioselectivity (4:1) | |
| Dimethylamino | Good Yield | Very High Regioselectivity (>10:1) | |
| Styrenyl Side (Alkyl) | 2-Me, 3-Me, 4-Me | 49-82 | 54:46 to 86:14 |
| Styrenyl Side (Alkoxy) | 2-OMe, 3-OMe, 4-OMe | 62-90 | 59:41 to 78:22 |
| Styrenyl Side (Halo) | 4-F | 13 | 85:15 |
| Styrenyl Side (Trisubstituted Alkene) | Alkyl substituents | Accepted | Not Specified |
| Styrenyl Side (Bulky) | 2-Naphthyl | 63 | 52:48 (Racemic) |
| N-Methylated Aniline | N-Me | 72 | 72:28 |
Experimental Protocols
Protocol 1: Photoenzymatic Enantioselective Synthesis of α-Fluorinated Amides
This protocol describes the synthesis of fluorinated amides with distal chirality using a visible-light-driven ene-reductase system [5].
Reagents and Materials
- Enzyme: OYE1 ene-reductase (wild-type or engineered Y375F variant)
- Cofactor Regeneration System: Glucose dehydrogenase (GDH), D-(+)-glucose, NADP+
- Fluorinated Substrate: Bromodifluoroacetamide derivative (e.g., F-1)
- Alkene Substrate: Electron-rich alkenes
- Buffer: Tris-HCl buffer (100 mM, pH 8.5)
- Light Source: 45 W blue LED (465 nm)
Step-by-Step Procedure
- Reaction Setup: In a glass vial, combine the bromodifluoroacetamide substrate F-1 (0.1 mmol), alkene (0.12 mmol), OYE1 enzyme (2 mol%), GDH (2 mg), NADP+ (1 mg), and D-(+)-glucose (10 mg) in 2 mL of Tris-HCl buffer (100 mM, pH 8.5).
- Photoreaction: Seal the vial and place it under illumination with a 45 W blue LED (465 nm) with constant stirring. Maintain the reaction temperature at 25°C.
- Reaction Monitoring: Monitor the reaction progress by GC-MS or TLC. Typical reaction time is 24-48 hours.
- Work-up: After completion, extract the reaction mixture with ethyl acetate (3 × 5 mL). Combine the organic layers and dry over anhydrous Na₂SO₄.
- Purification: Concentrate the organic extract under reduced pressure and purify the crude product by flash chromatography on silica gel to obtain the desired α-fluorinated amide.
Key Experimental Observations and Tips
- Enzyme Engineering: The Y375F mutation in OYE1 was critical, increasing yield to >90% while maintaining stereoselectivity (>90% ee) [5].
- Control Experiments: No product formation was observed in the absence of either the ER enzyme or blue light illumination [5].
- Substrate Specificity: The amide substrate (F-1) showed successful product formation, while the ester analogue (ethyl bromodifluoroacetate, F-0) yielded no product, highlighting the importance of the amide moiety for binding and reactivity [5].
Protocol 2: Stereoselective Photoenzymatic Hydroarylation for Quaternary Carbon Centers
This protocol details the use of an engineered Baeyer-Villiger Monooxygenase (AcHYAR) for the synthesis of tetrahydroquinolines via hydroarylation [32].
Reagents and Materials
- Enzyme: Engineered AcHYAR variant (AcCHMO-M10-R327K-R490E-I491E-Y246F-S186W-T187V)
- Substrate: Aniline derivative bearing a pendant 1,1-disubstituted alkene (e.g., compound 1)
- Buffer: Potassium phosphate buffer (50 mM, pH 7.5)
- Light Source: Blue LED array
Step-by-Step Procedure
- Enzyme Preparation: Express and purify the AcHYAR enzyme variant using standard protein expression techniques.
- Reaction Setup: In a reaction vial, combine the aniline substrate (0.05 mmol) and the AcHYAR enzyme (5 mg) in 1 mL of potassium phosphate buffer (50 mM, pH 7.5).
- Photoreaction: Seal the vial and irradiate with a blue LED array with constant stirring at 25°C for 24 hours.
- Reaction Monitoring: Monitor the reaction by LC-MS or TLC.
- Work-up and Purification: Extract the reaction mixture with ethyl acetate (3 × 3 mL). Dry the combined organic layers over Na₂SO₄, concentrate, and purify the product by preparative TLC or flash chromatography.
Key Experimental Observations and Tips
- Directed Evolution: The high performance of the AcHYAR enzyme was achieved through three rounds of iterative site-saturation mutagenesis, focusing on residues near the FAD cofactor [32].
- Structural Insight: A key disulfide bond between residues C326 and C489 in AcHYAR is crucial for activity and selectivity; mutating these cysteines to alanine significantly reduced yield and enantioselectivity [32].
- Scope Limitation: Electron-deficient substituents on the styrenyl side (e.g., trifluoromethyl) were not tolerated and led to alkylation of the flavin cofactor [32].
Workflow and Pathway Diagrams
The following diagrams illustrate the general workflow for photoenzymatic catalysis and the specific mechanism for the hydroarylation reaction.
Photoenzymatic Reaction Workflow
Hydroarylation Mechanism
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Photoenzymatic Synthesis
| Reagent / Material | Function / Role | Example & Key Detail |
|---|---|---|
| Ene-Reductases (OYE1) | Catalyzes enantioselective radical hydroalkylation of alkenes [5]. | OYE1; Y375F mutation boosts yield to >90% [5]. |
| Engineered BVMO (AcHYAR) | Catalyzes enantioselective hydroarylation for quaternary stereocenters [32]. | AcCHMO-M10-R327K-R490E-I491E-Y246F-S186W-T187V [32]. |
| Glucose Dehydrogenase (GDH) | Regenerates NADPH cofactor for sustained catalysis [5]. | Used with D-(+)-glucose as a sacrificial electron donor [5]. |
| Bromodifluoroacetamide | Radical precursor for introducing fluorinated motifs [5]. | Substrate F-1; amide group is crucial for enzyme binding [5]. |
| Flavin Cofactors (FAD/FMN) | Serve as intrinsic photoactive chromophores in flavoproteins [5] [32]. | FAD in AcHYAR; absorbs blue light to initiate radical processes [32]. |
| Blue LED Light Source | Provides photoexcitation energy for radical generation [5] [32]. | 45 W, 465 nm; optimal power/wavelength for excitation [5]. |
Optimizing Photoenzymatic Systems: Engineering and Reaction Design
Photoenzymatic synthesis merges the precise selectivity of enzymes with the unique reactivity unlocked by light, creating a powerful tool for asymmetric synthesis. The efficiency and success of these reactions are profoundly influenced by key reaction parameters, including the choice of buffer, pH, and light wavelength. This application note provides a consolidated guide to these critical parameters, equipping researchers with practical data and protocols to optimize photoenzymatic reactions for the synthesis of high-value chiral intermediates.
Summarized Parameter Effects and Quantitative Data
The following tables consolidate key quantitative findings on how buffer, pH, and light wavelength impact various photoenzymatic systems.
Table 1: Summary of Light Wavelength and Intensity Effects on Photoenzyme Activity
| Enzyme | Optimal Wavelength | Compared Wavelength | Observed Effect | Reference |
|---|---|---|---|---|
| Fatty Acid Photodecarboxylase (CvFAP) | Violet (~395 nm), Low Intensity/Pulsed | Blue Light | ~6-fold higher activity; reduced photoinactivation [33] | |
| Alpha-Amylase | 589 nm (Yellow), 20 J/cm² | 532 nm (Green) | Activity increased to 120% of control [34] | |
| Alpha-Amylase | 532 nm (Green), 40 J/cm² | 589 nm (Yellow) | Activity inhibited to 75% of control [34] | |
| Ene-Reductase (for fluorinated amides) | 465 nm, 45 W | 455 nm, 12 W | Yield increased from 15% to 21% [35] |
Table 2: Summary of Buffer and pH Effects on Photoenzymatic Reactions
| Enzyme / System | Optimal Buffer & pH | Comparison | Observed Effect | Reference |
|---|---|---|---|---|
| Ene-Reductase (OYE1) | Tris-HCl, pH 8.5 | Sodium Phosphate Buffer | Yield increased by ~1.7 times [35] | |
| Cross-linked Aldo-Keto Reductase (AKR) | Phosphate Buffer, pH 7.0 | Not Specified | Standard condition for synthesis of (R)-3,5-BTPE [36] | |
| Ketoreductase (LkKRED) | Potassium Phosphate Buffer (KPB), pH 7.0 | Not Specified | Standard condition for one-pot cascade reaction [23] |
Detailed Experimental Protocols
Protocol: Investigating Wavelength Effects on Fatty Acid Photodecarboxylase (FAP)
This protocol is adapted from studies investigating the enhancement of FAP activity using violet light [33].
I. Materials
- Enzyme: Purified CvFAP (expressed in E. coli and purified via Ni-NTA chromatography).
- Substrate: Palmitic acid (or other fatty acid of interest).
- Solvent: Degassed Tris-HCl buffer (100 mM, pH 8.0) with 30% DMSO.
- Light Sources: High-power LEDs emitting violet (~395 nm) and blue (~450-470 nm) light, calibrated with a power meter.
- Equipment: Stopped-flow spectrometer or suitable photobioreactor, anaerobic glovebox (N₂ atmosphere), UV-Vis spectrophotometer.
II. Method
- Sample Preparation:
- Prepare a 10 mM stock solution of palmitic acid in DMSO.
- In an anaerobic glovebox, prepare the final assay mix containing degassed Tris-HCl buffer, CvFAP, and palmitic acid substrate. The final DMSO concentration should be 30%.
Reaction Setup:
- Load the assay mix into the stopped-flow spectrometer or a sealed cuvette within the photobioreactor.
- Illuminate the reaction mixture using pre-calibrated violet or blue LEDs. For pulsed light experiments, use a programmable controller (e.g., 1 s on/1 s off cycles).
Activity Analysis:
- Real-Time CO₂ Detection: Monitor the reaction in real-time using a coupled assay that detects CO₂/bicarbonate formation by tracking NADH consumption at 340 nm.
- Time-Point Analysis: Alternatively, take aliquots at regular intervals. Stop the reaction and analyze for alkane product formation using GC or GC-MS.
III. Key Parameters
- Maintain a constant temperature (e.g., 25°C).
- Use a light intensity of 50 W for violet and blue LEDs for direct comparison.
- Compare initial rates of product formation or CO₂ release under violet vs. blue light illumination.
Protocol: Optimizing Buffer and pH for an Ene-Reductase Photo-Biocatalytic Reaction
This protocol is based on the optimization of a system for synthesizing fluorinated amides [35].
I. Materials
- Biocatalyst: Ene-reductase (e.g., OYE1, whole cells or purified enzyme).
- Cofactor Regeneration System: Glucose dehydrogenase (GDH), D-(+)-glucose, NADP⁺.
- Substrates: Fluorinated bromoamide (e.g., F-1) and electron-rich alkene.
- Buffers: 50 mM Sodium Phosphate (NaPi) and Tris-HCl buffers across a pH range (e.g., pH 6.5, 7.5, 8.5).
- Light Source: Blue LED (465 nm, 45 W).
- Equipment: Schlenk tubes or vials, photoirradiation setup, GC-MS or HPLC for analysis.
II. Method
- Reaction Setup:
- In a reaction vial, prepare the following mixture:
- Substrate F-1 (0.1 mmol)
- Alkene (0.12 mmol)
- Ene-reductase (2 mol%)
- GDH (2 mg), NADP⁺ (1 mg), D-glucose (5 equiv)
- Buffer (1 mL, 50 mM) of varying composition and pH.
- Purge the reaction vial with an inert gas (N₂ or Ar).
- In a reaction vial, prepare the following mixture:
Photo-Biocatalytic Reaction:
- Irradiate the reaction mixture with constant stirring under a 45 W blue LED (465 nm) for the specified duration (e.g., 24 h).
- Maintain the reaction temperature at a constant level (e.g., 30°C).
Product Analysis:
- Quench the reaction by extraction with an organic solvent (e.g., ethyl acetate).
- Analyze the organic extract by GC-MS or chiral HPLC to determine conversion and enantiomeric excess (ee).
III. Key Parameters
- Test both Tris-HCl and Sodium Phosphate buffers at identical pH values (e.g., 7.5 and 8.5) to directly compare buffer effects.
- Systematically vary the pH in each buffer system to identify the optimal pH for activity and enantioselectivity.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Reagents and Materials for Photoenzymatic Synthesis
| Reagent/Material | Function in Photoenzymatic Synthesis | Example Application |
|---|---|---|
| Tris-HCl Buffer | Maintains alkaline pH; often superior for activity in certain photo-enzymatic systems. | Optimal buffer for ene-reductase OYE1, significantly boosting yield [35]. |
| Potassium Phosphate Buffer (KPB) | Maintains neutral pH; a standard, compatible buffer for many enzymes. | Used in ketoreductase (LkKRED) cascades [23]. |
| Violet LED (~395 nm) | Excites flavin cofactors, can lead to higher catalytic efficiency and reduced photoinactivation. | 6-fold higher activity for FAP compared to blue light [33]. |
| Blue LED (455-465 nm) | Standard wavelength for exciting flavin-dependent enzymes (e.g., Ene-reductases, FAP). | Driving radical generation for enantioselective hydroalkylation [35]. |
| rGQDs/AKR Hybrid Catalyst | Cofactor-independent photo-biocatalyst; uses water as hydride source under IR light. | Synthesis of (R)-3,5-BTPE with >99.99% ee [36]. |
| Immobilized Catalysts (e.g., BCL@DON) | Facilitates catalyst recycling and improves stability, enabling continuous-flow processes. | Chemoenzymatic synthesis of chiral β-nitroalcohols in flow [37]. |
Visualized Workflows and Signaling Pathways
The following diagrams illustrate the core concepts and experimental workflows discussed in this note.
Diagram 1: A general workflow for systematically optimizing key parameters in photoenzymatic synthesis, highlighting critical decision points for light wavelength and buffer/pH selection.
Diagram 2: The catalytic mechanism of Fatty Acid Photodecarboxylase (FAP), comparing the enhanced reaction pathway initiated by violet light with the standard blue light pathway.
Strategies for Directed Evolution and Enzyme Engineering
Directed evolution and enzyme engineering are indispensable disciplines for advancing photoenzymatic enantioselective synthesis, a field that merges the precision of biocatalysis with the transformative power of photochemistry. Natural enzymes, while highly efficient under physiological conditions, often lack the robustness, specificity, or catalytic versatility required for industrial synthetic pathways, particularly in novel photo-driven reactions [38]. The core objective of enzyme engineering is to bridge this performance gap. By employing strategies that mimic natural selection in the laboratory, researchers can create tailor-made enzymes with enhanced properties, thereby enabling more efficient, sustainable, and stereoselective synthesis of complex molecules, such as pharmaceutical intermediates [39]. This document outlines the primary strategies—directed evolution, rational/semi-rational design, and machine learning-aided engineering—framed within the context of photoenzymatic research. It provides detailed application notes and protocols to equip scientists with the tools for developing next-generation biocatalysts.
Foundational Strategies and Their Applications
The engineering of enzymes relies on a spectrum of methodologies, from purely random approaches to computationally driven designs. The table below summarizes the core strategies, their key principles, and primary outputs.
Table 1: Core Strategies in Directed Evolution and Enzyme Engineering
| Strategy | Key Principle | Primary Output/Metric | Typical Experimental Workflow |
|---|---|---|---|
| Directed Evolution [40] [39] | Introduction of random mutations followed by high-throughput screening for desired traits. | Variants with improved thermostability, activity, or expression. | Gene Diversification → Expression → Screening/Selection → Iteration |
| Semi-Rational Design (Co-MdVS) [38] | Identification of co-evolving residue pairs via sequence analysis, followed by multidimensional computational screening of mutants. | Stabilized mutants with longer half-life ((t{1/2})) and improved catalytic efficiency ((k{cat}/K_M)). | Coevolutionary Analysis → Library Design → Multidimensional Virtual Screening → Experimental Validation |
| Machine Learning (ML)-Aided Design [41] | Use of algorithms to predict enzyme function, stability, and activity from sequence and structural data. | Models for predicting EC number, catalytic residues, and optimal mutation combinations. | Data Curation → Model Training → Prediction → Experimental Testing |
Directed Evolution: A Darwinian Approach in the Laboratory
Directed evolution is a powerful iterative process that simulates natural selection to engineer improved enzymes. Its major strength lies in its ability to discover beneficial mutations without requiring prior structural knowledge of the protein [39]. The process is exemplified by the engineering of nattokinase for enhanced fibrinolytic activity and the development of DNA polymerases (e.g., KAPA HiFi) for superior performance in Next-Generation Sequencing (NGS) workflows [40] [38]. A key application in synthetic biology includes the evolution of antibody mimics, such as fibronectin type III domain (10Fn3) scaffolds, which can be selected for high-affinity binding to therapeutic targets like TNF-α, achieving picomolar dissociation constants ((K_d)) [42].
Figure 1: Directed Evolution Workflow.
Protocol 1: Basic Directed Evolution Cycle
- Objective: To generate an enzyme variant with enhanced thermostability.
- Materials:
- Plasmid containing the gene of interest.
- E. coli JM109 or similar expression strain.
- PrimeSTAR Max DNA Polymerase (Takara) or other mutagenic PCR reagents.
- Luria-Bertani (LB) and 2x YT media.
- Selection antibiotics (e.g., Ampicillin, Kanamycin).
- Substrate for activity assay (e.g., Suc-AAPF-pNA for proteases [38]).
- Procedure:
- Gene Diversification: Create a library of gene variants. This can be achieved through error-prone PCR or by using commercial mutagenesis kits. The goal is to generate a library of millions of unique genes, each coding for a slightly different enzyme [40].
- Library Construction: Clone the diversified gene pool into an appropriate expression vector. Transform the constructs into a microbial host (e.g., E. coli) to create a library of variants.
- Expression and Screening: Culture the transformants, ideally in a high-throughput format (e.g., in 96-well plates). Induce protein expression. Apply a functional selection pressure, such as incubating at an elevated temperature, to identify thermostable variants. Screen for retained activity using a colorimetric or fluorogenic assay [40] [38].
- Selection and Iteration: Isolate the genes from the best-performing clones (e.g., those showing highest activity after heat treatment). Use these as the template for the next round of diversification and selection. Repeat the cycle until the desired stability is achieved [39].
Semi-Rational Design: The Co-MdVS Strategy for Enhanced Robustness
Semi-rational strategies combine evolutionary principles with computational power to reduce the experimental workload and address complex traits like stability. The Co-MdVS (Coevolutionary analysis and Multidimensional Virtual Screening) strategy is a prime example, successfully applied to nattokinase (NK) [38]. This approach leverages the insight that co-evolving residues in protein sequences are critical for stability.
Table 2: Key Outcomes from the Co-MdVS Strategy Applied to Nattokinase [38]
| Parameter | Wild-Type NK | Evolved Mutant M6 | Fold Improvement |
|---|---|---|---|
| Half-life at 55°C ((t_{1/2})) | Baseline | 31-fold increase | 31x |
| Acid Resistance | Low | Significantly enhanced | Qualitative |
| Catalytic Efficiency ((k{cat}/KM)) | Baseline | Improved with different substrates | Variable |
| Number of Mutations | - | 6 (combined from iterative cycles) | - |
Protocol 2: Implementing the Co-MdVS Strategy
- Objective: To design and screen for enzyme variants with significantly improved robustness using computational pre-screening.
- Materials:
- Multiple sequence alignment of the target enzyme family.
- Coevolutionary analysis software (e.g., EVcouplings).
- Molecular dynamics (MD) simulation software (e.g., GROMACS).
- Folding free energy (ΔΔG) calculation tools (e.g., FoldX).
- PrimeSTAR Max DNA Polymerase and reagents for site-directed mutagenesis.
- Procedure:
- Coevolutionary Analysis: Perform a deep sequence alignment of homologous proteins. Identify pairs of residues that show strong coevolutionary signals. These pairs are potential hotspots for stabilizing combinatorial mutations [38].
- Virtual Library Construction: Design a virtual mutation library focusing on the identified co-evolving residue pairs. For example, from 7980 possible dual mutants for nattokinase [38].
- Multidimensional Virtual Screening (MdVS): Screen the library using multiple computational metrics:
- Energetic Stability: Calculate the change in folding free energy (ΔΔG). Prefer mutants with negative ΔΔG, indicating stabilized folding.
- Structural Dynamics: Run short MD simulations. Calculate the root mean square deviation (RMSD), radius of gyration (Rg), and total number of hydrogen bonds. Select mutants with lower RMSD, lower Rg, and more hydrogen bonds than the wild-type [38].
- Experimental Validation: Synthesize the top-ranked candidates (e.g., 8 dual mutants) using site-directed mutagenesis [38]. Express and purify the proteins. Validate robustness by measuring half-life at target temperatures and catalytic efficiency.
Figure 2: Co-MdVS Design Workflow.
Machine Learning in Enzyme Engineering
Machine learning (ML) is revolutionizing enzyme engineering by enabling the prediction of function and optimization of properties from vast datasets. ML models can predict Enzyme Commission (EC) numbers from sequence alone (e.g., DeepEC, ECPred), identify catalytic residues (e.g., PREvaIL), and predict optimal experimental conditions like temperature and solubility [41]. This is particularly valuable for navigating the immense sequence space and identifying non-obvious, beneficial mutations.
Protocol 3: Leveraging ML for Enzyme Property Prediction
- Objective: To use pre-trained ML models to predict the function and optimal growth temperature of a novel enzyme sequence.
- Materials:
- Amino acid sequence of the enzyme in FASTA format.
- Access to ML prediction webservers or downloadable tools (see Table I in [41]).
- Procedure:
- Function Prediction: Submit your protein sequence to a tool like DeepEC or ECPred. These tools use convolutional neural networks or ensemble methods to predict the complete or partial EC number, providing a first-pass functional annotation [41].
- Condition Optimization: Use a tool like TOME or TAXyl, which are random forest-based classifiers, to predict the optimal growth and catalytic temperature of the enzyme based on its sequence, helping to guide experimental design [41].
- Solubility Prediction: Assess the potential for soluble expression in E. coli using a tool like SoluProt prior to cloning, saving time and resources [41].
Application in Photoenzymatic Enantioselective Synthesis
The engineered enzymes produced by these strategies are pivotal for advanced synthesis. A seminal application is a one-pot photo-enzymatic cascade process for the enantioselective C(sp³)–H functionalization of saturated N-heterocycles, such as phenylpyrrolidine—a key pharmaceutical building block [22]. This innovative method integrates a light-driven C–N cross-coupling reaction (using a dual Ni/photocatalyst system in DMSO) with a biocatalytic carbene transfer step catalyzed by an engineered SD-VHbCH carbene transferase within a whole-cell system [22]. The engineered enzyme provides a stable reaction environment that is critical for achieving superior stereoselectivity (up to 99% ee) in the final functionalized product, a feat difficult to accomplish with traditional chemical catalysts alone [22].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Directed Evolution and Enzyme Engineering
| Reagent / Kit | Function / Application | Key Feature |
|---|---|---|
| KAPA HiFi DNA Polymerase [40] | High-fidelity PCR for library amplification and mutagenesis. | Engineered via directed evolution for ultra-high fidelity and robustness in NGS library prep. |
| KAPA HyperPrep Kit [40] | Efficient construction of sequencing-ready libraries. | Provides high library yields, reduced duplicates, and improved coverage depth. |
| KAPA RNA HyperPrep Kit [40] | RNA library preparation for transcriptomic studies. | Enables flexible workflows for selective mRNA capture or rRNA depletion. |
| Suc-AAPF-pNA [38] | Chromogenic substrate for protease activity assays (e.g., nattokinase). | Allows direct spectrophotometric measurement of enzyme activity at 405 nm. |
| PrimeSTAR Max DNA Polymerase [38] | High-performance PCR for site-directed mutagenesis. | Used for efficient amplification in full-length plasmid PCR for mutant construction. |
| Ni/PC Photocatalyst System [22] | Light-driven C–N cross-coupling in photo-enzymatic cascades. | Enables the initial coupling of aryl bromides with cyclic amines under blue LED irradiation. |
| Engineered Carbene Transferase (e.g., SD-VHbCH) [22] | Biocatalytic enantioselective functionalization in cascade reactions. | Provides a stable binding pocket for high enantioselectivity (up to 99% ee) in synthesis. |
Photoenzymatic catalysis represents a groundbreaking fusion of photocatalysis and enzymatic catalysis, combining the high reactivity from photoexcitation with the exceptional stereoselectivity inherent to biocatalysis [1]. This interdisciplinary approach has created exciting opportunities for tackling long-standing challenges in enantioselective radical reactions and accessing new-to-nature enzyme reactivities. However, researchers frequently encounter two significant limitations: low reaction yield and poor enantioselectivity. These constraints substantially hinder the practical application of photoenzymatic systems in pharmaceutical development and fine chemical synthesis, where both efficiency and stereochemical purity are paramount.
The fundamental challenge stems from managing highly reactive radical intermediates while maintaining precise stereochemical control within enzyme active sites. Radical species, while offering valuable synthetic versatility, often participate in unproductive side reactions that diminish yield. Simultaneously, the enzyme's chiral environment must effectively discriminate between competing transition states to achieve high enantioselectivity. This application note details targeted strategies to overcome these limitations through systematic reaction optimization and enzyme engineering, with a specific focus on protocols relevant to drug development applications.
Strategic Approaches and Experimental Protocols
Systematic Reaction Optimization Framework
Initial reaction optimization begins with fundamental parameters that profoundly influence both yield and enantioselectivity. The provided case studies demonstrate that meticulous adjustment of these variables can dramatically enhance performance.
Light Source and Wavelength Control: Precise management of irradiation parameters is crucial. In the synthesis of fluorinated amides, switching from a 12 W 455 nm to a 45 W 465 nm light source increased yields from 15% to 21% while maintaining excellent enantioselectivity (93% ee) [5]. This improvement was attributed to more efficient substrate excitation and superior penetration through the reaction mixture.
Buffer and pH Optimization: The choice of buffer system significantly impacts catalytic efficiency. Comparative studies between sodium phosphate (NaPi) and Tris-HCl buffers across various pH levels revealed superior performance in alkaline conditions. Transitioning from NaPi (pH 7.0) to Tris-HCl (pH 8.5) increased reaction yield by approximately 1.7-fold, indicating the critical influence of protonation states on the catalytic mechanism [5].
Enzyme and Cofactor Loading: Strategic increases in enzyme loading can substantially improve yield without compromising selectivity. Elevating ene-reductase loading from 1 mol% to 2 mol% boosted yield from 21% to 51% while preserving 93% enantiomeric excess [5]. Similarly, ensuring adequate concentrations of the cofactor regeneration system (NADP+, glucose dehydrogenase, glucose) is essential for sustaining catalytic turnover.
Table 1: Reaction Optimization Parameters and Performance Outcomes
| Parameter | Initial Condition | Optimized Condition | Impact on Yield | Impact on ee |
|---|---|---|---|---|
| Light Source | 12 W, 455 nm | 45 W, 465 nm | 15% → 21% | Unchanged (93%) |
| Buffer System | NaPi, pH 7.0 | Tris-HCl, pH 8.5 | 1.7-fold increase | Maintained >90% |
| Enzyme Loading | 1 mol% | 2 mol% | 21% → 51% | Unchanged (93%) |
| Temperature | Room temperature | -20°C (with MS) | Variable by system | Improved (e.g., 82:18 → 90:10 e.r.) [43] |
Enzyme Engineering for Enhanced Performance
When reaction optimization reaches its limits, enzyme engineering provides a powerful strategy for achieving breakthrough improvements. The following protocol outlines a structured approach to enzyme engineering through site-directed mutagenesis and directed evolution.
Protocol: Site-Directed Mutagenesis for Photoenzyme Enhancement
Objective: Identify and modify key amino acid residues to improve enzyme activity and stereocontrol.
Materials:
- Wild-type enzyme (e.g., OYE1, XenB)
- Site-directed mutagenesis kit
- Oligonucleotide primers for targeted mutations
- Expression system (E. coli or other suitable host)
- Chromatography equipment for protein purification
Procedure:
- Identify Target Residues: Perform molecular docking studies with substrates and intermediates to identify residues within 5-7 Å of the binding pocket. For OYE1, key residues included Y375, Y196, F123, W116, Y82, M39, and T37 [5].
- Design Mutations: Create a focused mutant library targeting these positions with amino acid substitutions that modulate steric bulk and electronic properties (e.g., alanine for reduced size, phenylalanine for maintained aromaticity with altered geometry).
- Generate Mutants: Implement site-directed mutagenesis using appropriate molecular biology techniques.
- Express and Purify: Express variant enzymes in a suitable host system and purify using affinity chromatography.
- High-Throughput Screening: Screen variants for both activity and enantioselectivity using GC-MS or HPLC analysis.
Implementation Example: In fluorinated amide synthesis, the Y375F mutation in OYE1 increased yield from 51% to >90% while maintaining excellent stereoselectivity (>90% ee) [5]. Computational analysis revealed this mutation enhanced binding affinity for the radical intermediate, facilitating more efficient catalysis.
Protocol: Directed Evolution for Radical Hydroamination
Objective: Evolve enzyme variants with improved activity and selectivity for challenging radical reactions.
Materials:
- Parent enzyme (e.g., XenB from Pseudomonas putida)
- Error-prone PCR or DNA shuffling materials
- High-throughput screening platform
- NADP+, glucose dehydrogenase, glucose for cofactor regeneration
- Analytical equipment (GC, HPLC, LC-MS)
Procedure:
- Library Creation: Generate genetic diversity through error-prone PCR or other mutagenesis methods.
- Primary Screening: Assess variants for improved activity using colorimetric or fluorometric assays.
- Secondary Screening: Evaluate promising variants for enantioselectivity using chiral chromatography.
- Iterative Rounds: Perform additional rounds of mutagenesis and screening to accumulate beneficial mutations.
- Characterization: Purify top-performing variants and determine kinetic parameters and enantioselectivity under standard conditions.
Implementation Example: Directed evolution of XenB for intermolecular radical hydroamination identified multiple beneficial mutations. The W100L variant (E1) provided 77% yield and 95% ee, while the A232L variant (E2) achieved 88% yield and 93% ee, substantially improving upon the wild-type performance [3].
Diagram 1: Enzyme Engineering Workflow - A strategic pathway for improving photoenzyme performance through reaction optimization and protein engineering.
Cofactor and Additive Engineering
The strategic use of additives and cofactor optimization can significantly enhance photoenzymatic systems by stabilizing radical intermediates and improving chiral induction.
Quaternary Ammonium Salts: Addition of nBu₄NCl (tetrabutylammonium chloride) in Al–salen photocatalytic systems improved yield from 44% to 67% while enhancing enantiomeric ratios from 74:26 to 90:10 for certain diastereomers [43]. This effect is attributed to stabilization of the excited-state catalyst-substrate complex.
Molecular Sieves: Incorporating molecular sieves (3Å or 4Å) in reactions conducted at reduced temperatures (-20°C) improved yields without compromising enantioselectivity by removing trace water that can interfere with radical propagation [43].
Case Studies in Pharmaceutical Synthesis
Synthesis of Fluorinated Amides with Remote Chirality
The development of visible-light-driven ene-reductase systems for synthesizing fluorinated amides demonstrates the power of integrated optimization approaches. Fluorinated compounds are increasingly important in pharmaceutical development, with fluorine-containing drugs now exceeding 50% of all pharmaceuticals, yet their enantioselective synthesis remains challenging [5].
Table 2: Performance of Optimized Photoenzymatic Systems
| Reaction Type | Enzyme | Optimized Yield | Optimized ee | Key Optimization |
|---|---|---|---|---|
| Fluorinated Amide Synthesis | OYE1 (Y375F) | Up to 91% | Up to 97% | Enzyme engineering, light optimization [5] |
| Intermolecular Hydroamination | XenB (A232L) | Up to 88% | 93-97% | Directed evolution, buffer optimization [3] |
| Photocyclization | Al–salen Al-1 | 67% | 90:10 e.r. (cis) | Additives (nBu₄NCl), temperature control [43] |
Substrate Design Strategy: Critical to success was the strategic design of fluorinated precursors. While ethyl bromodifluoroacetate (F-0) failed to produce products under blue light irradiation, replacing the ester with an amide group (F-1) enabled successful conversion. Computational studies revealed the amide substrate formed stronger binding interactions with the enzyme active site, highlighting the importance of substrate-enzyme complementarity [5].
Scope and Limitations: The optimized system tolerated diverse functional groups including pharmaceutically relevant cyclobutyl, chloro, trifluoromethyl, ether, and 1,3-dioxolane groups, producing compounds with 47-87% yield and 93-97% ee. However, aliphatic olefins such as 2-methylbutene showed poor reactivity, indicating limitations in substrate scope that may require additional enzyme engineering efforts [5].
Intermolecular Radical Hydroamination
The photoenzymatic production of nitrogen-centered radicals for enantioselective intermolecular hydroamination addresses a long-standing challenge in synthetic chemistry. Before this development, catalytic enantioselective intermolecular radical hydroamination remained elusive due to the short lifetime of nitrogen-centered radicals and competing side reactions [3].
Mechanistic Insight: The enantioselectivity originates from the radical-addition step within the enzyme's chiral environment, while the reactivity stems from ultrafast photoinduced electron transfer from reduced flavin mononucleotide (FMNH-) to nitrogen-containing substrates. This system operates efficiently at room temperature under visible light without external photocatalysts [3].
Scale-Up Potential: Gram-scale reactions demonstrated similar reactivity and selectivity to small-scale standard conditions, highlighting the potential for industrial application in producing chiral amine building blocks for pharmaceutical synthesis [3].
Diagram 2: Photoenzymatic Radical Mechanism - Key steps in the photoenzymatic catalytic cycle showing light absorption, electron transfer, and enantioselective bond formation.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of photoenzymatic methodologies requires specific reagents and materials optimized for radical chemistry and enzymatic stability.
Table 3: Essential Research Reagents for Photoenzymatic Synthesis
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Ene-reductases (OYE1, XenA/B) | Biocatalyst for radical reactions | Engineered variants show improved performance; requires coexpression with FMN cofactor [5] [3] |
| Flavin Mononucleotide (FMN) | Photoredox cofactor | Natural photoactive cofactor; cycles between oxidized and reduced states during catalysis [3] |
| NADP+/GDH/Glucose | Cofactor regeneration system | Maintains reducing equivalents for sustained catalytic turnover [5] |
| nBu₄NCl | Additive for stabilization | Enhances yield and enantioselectivity in photocyclization reactions [43] |
| Molecular Sieves (3Å/4Å) | Water scavenger | Critical for low-temperature photoenzymatic reactions [43] |
| Tris-HCl Buffer (pH 8.5) | Reaction medium | Optimal for many photoenzymatic systems; alkaline conditions improve efficiency [5] |
The integrated strategies presented in this application note provide a systematic framework for overcoming the persistent challenges of low yield and poor enantioselectivity in photoenzymatic synthesis. Through coordinated optimization of reaction parameters, strategic enzyme engineering, and thoughtful substrate design, researchers can achieve performance metrics suitable for pharmaceutical development. The case studies demonstrate that yield improvements from 15% to >90% and enantioselectivity exceeding 97% ee are attainable through these approaches.
Future developments in the field will likely focus on expanding the substrate scope of existing photoenzymatic systems through machine learning-guided protein design and the discovery of novel photoactive enzymes from diverse biological sources. As these methodologies mature, photoenzymatic catalysis is poised to become an indispensable tool for the sustainable synthesis of complex chiral molecules in pharmaceutical and fine chemical industries.
The Role of Synergistic Dual Catalysis with Organic Dyes
Synergistic dual catalysis, which merges the power of photoredox catalysis with the precision of enzymatic catalysis, has established itself as a transformative platform for asymmetric synthesis. This approach enables chemical transformations that are notoriously challenging to achieve using traditional catalytic methods alone [2]. By integrating the remarkable stereocontrol inherent to enzymes with the ability of photocatalysts to generate reactive intermediates under mild conditions, researchers can now access valuable chiral building blocks with unprecedented efficiency and selectivity.
Within this emerging field, organic dyes have proven particularly valuable as photocatalysts due to their biocompatibility, cost-effectiveness, and tunable redox properties. Unlike transition metal-based photocatalysts, organic dyes such as Rhodamine B operate effectively under visible light irradiation without causing enzyme denaturation, making them ideal partners for biocatalytic processes [4] [44]. This application note examines recent breakthroughs in photoenzymatic enantioselective synthesis, with a specific focus on methodologies enabling access to pharmaceutically relevant chiral amines and amino acids.
Key Advancements in Photoenzymatic Synthesis
Enantioselective Synthesis of Vicinal Diamines
Vicinal diamines represent privileged structural motifs found in numerous pharmaceuticals and biologically active molecules. Traditional synthetic approaches often struggle with stereocontrol when assembling these challenging scaffolds. A breakthrough solution employs a dual bio-/photo-catalytic system combining an ene-reductase (ERED) with the organic dye Rhodamine B under green light excitation [4].
This system achieves enantioselective hydroamination of enamides through the generation and control of nitrogen-centered radicals (NCRs). The process exhibits exceptional stereoselectivity, with reported enantiomeric excess values up to >99% ee [4]. The mechanism involves synergistic cooperation between the enzymatic and photocatalytic cycles: Rhodamine B generates NCRs from N-amidopyridinium salts under green light, while the engineered ene-reductase (GluER-M5) controls the stereoselective addition to the enamide substrate.
Table 1: Performance of Photoenzymatic Vicinal Diamine Synthesis
| Parameter | Optimized Performance | Key Factors |
|---|---|---|
| Enantioselectivity | Up to >99% ee | Engineered GluER-M5 variant |
| Yield | Up to 82% | Rhodamine B photocatalyst |
| Light Source | 530-540 nm green LEDs | Improved enzyme compatibility |
| Enzyme Loading | As low as 1 mol% | Glucose/GDH recycling system |
| Reaction Scale | 0.1 mmol demonstrated | Scalable with maintained selectivity |
Synthesis of Fluorinated Amides with Remote Stereocontrol
The incorporation of fluorine atoms into organic compounds represents a crucial strategy in modern drug design, with over 50% of recent pharmaceuticals containing fluorine [5]. Photoenzymatic approaches have now enabled the synthesis of fluorinated amides with distal chirality (γ-to F), a challenging stereochemical arrangement for conventional methods.
This transformation utilizes visible-light-driven ene-reductase systems to generate carbon-centered radicals from fluorine-containing brominated amides, followed by enantioselective hydroalkylation with alkenes [5]. Through enzyme engineering and reaction optimization, researchers achieved high yields (up to 91%) and excellent stereocontrol (up to 97% ee) for these valuable fluorinated building blocks.
Radical Synthesis of Non-Canonical Amino Acids
The synergistic merger of photoredox catalysis and pyridoxal 5'-phosphate (PLP) biocatalysis has enabled the development of a fundamentally new form of radical pyridoxal biocatalysis [44]. This approach allows the protecting-group-free preparation of valuable non-canonical amino acids, including those bearing stereochemical dyads or triads.
Notably, this system provides stereodivergent access to both enantiomeric products through biocatalyst-controlled editing of α-stereochemistry. Using engineered PLP enzymes, specifically the "2B9" variant of the Pyrococcus furiosus tryptophan synthase β-subunit (l-PfPLPβ), in combination with Rhodamine B as an organic photoredox catalyst, this method achieves C-C coupling with excellent enantiomeric ratios (93:7 e.r.) [44].
Experimental Protocols
General Workflow for Photoenzymatic Dual Catalysis
The following workflow illustrates the typical setup and execution of synergistic photoenzymatic reactions employing organic dyes:
Reagents and Materials
- N-(1-phenylvinyl)-acetamide (1a) (0.1 mmol, 1.0 equiv)
- N-amidopyridinium salt (2a) (0.12 mmol, 1.2 equiv)
- Engineered ene-reductase GluER-T36A-Y177F-F269V-K277M-Y343F (ER-M5) (1 mol%)
- Rhodamine B (10 mol%)
- Glucose dehydrogenase (GDH, 5 mg/mL)
- NADP+ (0.5 mM)
- D-glucose (10 mM)
- Tris-HCl buffer (50 mM, pH 8.5)
- Green LEDs (530-540 nm)
Step-by-Step Procedure
Reaction Setup: In a 5 mL glass vial equipped with a magnetic stir bar, combine N-(1-phenylvinyl)-acetamide (1a, 0.1 mmol) and N-amidopyridinium salt (2a, 0.12 mmol) in Tris-HCl buffer (2 mL, 50 mM, pH 8.5).
Catalyst Addition: Add sequentially the engineered ene-reductase ER-M5 (1 mol%), Rhodamine B (10 mol%), glucose dehydrogenase (5 mg/mL), NADP+ (0.5 mM), and D-glucose (10 mM).
Oxygen Exclusion: Seal the vial with a rubber septum and purge the headspace with nitrogen or argon for 10 minutes to remove dissolved oxygen, which can quench radical intermediates.
Photoreaction: Place the reaction vessel in a photoreactor equipped with green LEDs (530-540 nm) and illuminate with stirring at 25°C for 24-48 hours.
Reaction Monitoring: Monitor reaction progress by LC-MS or TLC. Typically, completion is observed within 24-48 hours.
Workup: Extract the reaction mixture with ethyl acetate (3 × 5 mL), combine the organic layers, dry over anhydrous Na₂SO₄, and concentrate under reduced pressure.
Purification: Purify the crude product by flash column chromatography on silica gel (hexane/ethyl acetate gradient) to afford the desired vicinal diamine product.
Analysis: Determine enantiomeric excess by chiral HPLC or SFC analysis. Characterize the product by ( ^1H ) NMR, ( ^{13}C ) NMR, and HRMS.
Reagents and Materials
- Serine or β-hydroxy-α-amino acid (0.1 mmol, 1.0 equiv)
- Benzyltrifluoroborate salt 1a (0.15 mmol, 1.5 equiv)
- Engineered l-PfPLPβ enzyme (2B9 variant) (1.0 mol%)
- Rhodamine B (10 mol%)
- Sodium phosphate buffer (50 mM, pH 6.0)
Step-by-Step Procedure
Reaction Setup: In a 5 mL glass vial, suspend the serine or β-hydroxy-α-amino acid substrate (0.1 mmol) in sodium phosphate buffer (2 mL, 50 mM, pH 6.0).
Catalyst System: Add the engineered l-PfPLPβ enzyme (1.0 mol%), benzyltrifluoroborate salt 1a (0.15 mmol), and Rhodamine B (10 mol%).
Degassing: Sparge the reaction mixture with nitrogen for 15 minutes to ensure complete oxygen removal.
Illumination: Irradiate the reaction with blue LEDs (450-455 nm) with continuous stirring at 25°C for 24-36 hours.
Monitoring: Track reaction progress by LC-MS. The consumption of the starting amino acid and formation of the non-canonical amino acid product should be observed.
Purification: After completion, centrifuge the reaction mixture to remove precipitated proteins. Purify the supernatant by preparative HPLC to isolate the non-canonical amino acid product.
Analysis: Determine enantiomeric ratio by chiral HPLC or by derivatization with Marfey's reagent followed by LC-MS analysis.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for Photoenzymatic Dual Catalysis
| Reagent/Catalyst | Function | Typical Loading | Notes |
|---|---|---|---|
| Rhodamine B | Organic photoredox catalyst | 10 mol% | Green light absorption; biocompatible |
| Ene-reductases (EREDs) | Stereoselective radical addition | 1-2 mol% | Engineered variants (e.g., GluER-M5) for improved performance |
| PLP-dependent enzymes | Amino acid radical biotransformation | 1.0 mol% | e.g., l-PfPLPβ (2B9 variant) for C-C coupling |
| GDH/Glucose/NADP+ | Cofactor recycling system | 5 mg/mL/10 mM/0.5 mM | Maintains enzymatic activity throughout reaction |
| N-Amidopyridinium salts | NCR precursors | 1.2 equiv | Generate N-centered radicals under green light |
| Fluorinated bromoamides | Fluorinated radical precursors | 1.5 equiv | Enable incorporation of F-atoms with stereocontrol |
| Alkyltrifluoroborates | Radical precursors for AA synthesis | 1.5 equiv | Stable, easily oxidized to corresponding radicals |
Mechanism and Synergistic Pathways
The remarkable efficiency of synergistic dual catalysis systems stems from the complementary roles of the organic dye photocatalyst and the enzyme. The following diagram illustrates the mechanistic pathway for the enantioselective synthesis of vicinal diamines:
The mechanism involves several key stages: (1) Photoexcitation of Rhodamine B generates an excited state capable of single-electron transfer; (2) Reduction of N-amidopyridinium salts triggers N-N bond cleavage, generating nitrogen-centered radicals; (3) These radicals add to enamide substrates pre-bound in the enzyme active site; (4) The flavin cofactor within the ene-reductase facilitates stereoselective hydrogen atom transfer (HAT), determining the final chiral configuration; (5) Product release regenerates the catalytic cycle [4].
Similar mechanistic principles apply to the photoenzymatic synthesis of fluorinated compounds and non-canonical amino acids, with the key distinction being the nature of the radical precursor and the enzyme class employed for stereocontrol [5] [44].
Application in Drug Development
The methodologies described herein offer significant advantages for pharmaceutical research and development. The ability to rapidly assemble complex chiral architectures, including vicinal diamines, fluorinated amides, and non-canonical amino acids, with exceptional stereocontrol addresses critical challenges in modern medicinal chemistry. These compounds serve as key precursors to pharmaceutical agents, peptide therapeutics, and functional biologics [5] [44].
The compatibility of these transformations with a range of functional groups, including heterocycles that might inactivate transition-metal catalysts, further enhances their utility in drug discovery campaigns [4]. Additionally, the mild reaction conditions (aqueous buffers, room temperature, visible light irradiation) and minimal protecting group manipulation align with green chemistry principles, making these approaches attractive for sustainable pharmaceutical manufacturing.
Synergistic dual catalysis combining organic dyes with enzymes represents a rapidly advancing frontier in asymmetric synthesis. The protocols and applications detailed in this document provide researchers with practical tools to implement these powerful methodologies. As enzyme engineering continues to expand the repertoire of compatible biotransformations and organic dye development enhances photocatalytic efficiency, this integrated approach promises to unlock new possibilities for the sustainable synthesis of complex chiral molecules with precision and efficiency.
Resolving Common Challenges with Radical Side Reactions
Radical intermediates offer immense potential for constructing valuable molecular architectures that are difficult to access via traditional two-electron pathways. However, their high reactivity and short lifetimes often lead to undesirable side reactions, presenting significant challenges in achieving controlled and selective transformations. Within the context of photoenzymatic enantioselective synthesis, researchers have developed innovative strategies to overcome these limitations by leveraging the unique properties of enzymatic systems.
This application note examines common radical side reactions and details experimental protocols for their suppression in photoenzymatic systems, enabling the synthesis of valuable chiral amines, fluorinated compounds, and heterocycles with excellent enantioselectivity.
Common Radical Side Reactions and Enzymatic Control Strategies
Table 1: Common Radical Side Reactions and Enzymatic Control Strategies
| Radical Side Reaction | Impact on Reaction | Enzymatic Control Strategy | Representative System |
|---|---|---|---|
| Hydrogen Atom Abstraction | Quenches radical, terminates catalytic cycle, reduces yield | Confined enzyme active site shields radical intermediate [3] | Ene-reductase-catalyzed hydroamination [3] |
| Uncontrolled Radical Generation | Leads to non-selective background reaction, poor stereocontrol | Optimization of light wavelength to match enzyme, not substrate [11] | Thioxanthone-based photoenzyme for [2+2] cycloaddition [11] |
| Radical Recombination/Dimerization | Forms undesired byproducts, consumes reactants | Pre-organization of substrates within active site favors productive pathways [3] | Ene-reductase-catalyzed hydroamination [3] |
| Competing Electron/Energy Transfer | Diverts reaction pathway, reduces efficiency | Genetic encoding of optimized sensitizers (e.g., thioxanthone) [11] | Engineered Methanococcus jannaschii tyrosyl-tRNA synthetase/tRNA pair [11] |
Nitrogen-centered radicals (NCRs) are particularly prone to hydrogen atom abstraction due to their high reactivity, which prematurely quenches the radical intermediate and terminates the catalytic cycle [3]. In the photoenzymatic hydroamination system using the ene-reductase XenB, the confined chiral environment of the enzyme's active site effectively shields the photogenerated nitrogen-centered radical, preventing this deleterious pathway and enabling enantioselective addition to alkenes [3].
Uncontrolled Radical Generation
A significant challenge in photochemical reactions is the uncontrolled direct excitation of substrates, which leads to non-selective background reactions. This was observed with a carbon-linked quinolone derivative, where irradiation at 365 nm caused a high background reaction [11]. The solution was engineering a photoenzyme incorporating a thioxanthone sensitizer, which allowed the use of longer wavelength visible light (>395 nm) that the substrate does not absorb, thus suppressing direct excitation and enabling selective energy transfer catalysis [11].
Experimental Protocols
Photoenzymatic Intermolecular Radical Hydroamination
This protocol describes an enantioselective intermolecular hydroamination between carbamate-functionalized amine precursors and alkenes, catalyzed by an engineered ene-reductase (XenB variant) [3].
Research Reagent Solutions
Table 2: Key Reagents for Photoenzymatic Hydroamination
| Reagent | Function | Specifications/Notes |
|---|---|---|
| Engineered Ene-reductase (XenB variant) | Biocatalyst | XenB-W100L or XenB-A232L provide high enantioselectivity [3] |
| Flavin Mononucleotide (FMN) | Enzyme cofactor | Natural photoactive cofactor within ene-reductases |
| NADP+ / Glucose Dehydrogenase (GDH) / D-Glucose | Cofactor regeneration system | Maintains FMN in its reduced state (FMNH-) for photoexcitation |
| Carbamate Substrate (e.g., 1a) | Nitrogen-centered radical precursor | Features N-O bond (e.g., 4-cyanophenolate leaving group) [3] |
| Sodium Phosphate (NaPi) or Imidazole Buffer | Reaction medium | pH is critical; optimal yield at pH 6.5 in imidazole buffer [3] |
Procedure
Reaction Setup: In a 4 mL clear glass vial, combine the following:
- Carbamate substrate (e.g., 1a, 0.05 mmol)
- Alkene substrate (e.g., α-methyl styrene, 0.10 mmol)
- Engineered XenB variant (e.g., E1 or E2, 5 mg)
- NADP+ (0.5 μmol)
- GDH (5 mg)
- D-Glucose (20 μmol)
- Imidazole buffer (2 mL, 50 mM, pH 6.5)
Photoreaction: Seal the vial and place it in a photoreactor equipped with blue light-emitting diodes (LEDs, λmax = 450 nm). Irradiate the reaction mixture for 12-24 hours at room temperature with constant stirring.
Monitoring: Monitor reaction progress by analytical techniques such as thin-layer chromatography (TLC) or gas chromatography (GC).
Work-up: After completion, extract the reaction mixture with ethyl acetate (3 × 5 mL). Combine the organic extracts and dry over anhydrous sodium sulfate.
Purification: Concentrate the organic layer under reduced pressure and purify the crude product by flash chromatography on silica gel to obtain the desired chiral amine product (e.g., 3a).
Troubleshooting Tips:
- Low Yield: Ensure the light source is functional and the vial is positioned for direct illumination. Verify the activity of the enzyme and cofactor regeneration system.
- Low Enantioselectivity: Confirm that the correct engineered enzyme variant (E1 or E2) is being used and check for potential enzyme denaturation.
Dual Photobiocatalytic System for Vicinal Diamine Synthesis
This protocol employs a synergistic approach using an ene-reductase (GluER mutant) and an organic photosensitizer (Rhodamine B) to achieve enantioselective hydroamination of enamines for the synthesis of chiral vicinal diamines [4].
Procedure
Reaction Setup: In a 4 mL glass vial, combine:
- Enamide substrate (e.g., 1a, 0.05 mmol)
- N-amidopyridinium salt (e.g., 2a, 0.10 mmol)
- Engineered GluER mutant (ER-M5, 1 mol%)
- Rhodamine B (RhB, 1 mol%)
- NADP+ (0.5 μmol)
- GDH (5 mg)
- D-Glucose (20 μmol)
- Phosphate buffer (2 mL, 50 mM, pH 7.0)
Photoreaction: Seal the vial and place it in a photoreactor equipped with green LEDs (λmax = 530 nm). Irradiate for 12-24 hours at room temperature with stirring.
Monitoring and Work-up: Monitor by TLC or LC-MS. Work up as described in Protocol 3.1.
Purification: Purify the crude material by flash chromatography to afford the vicinal diamine product (e.g., (S)-3a).
Troubleshooting Tips:
- No Reaction: Confirm the wavelength of the light source matches the absorption profile of Rhodamine B (green light). Ensure the enzyme and photocatalyst are properly stored and active.
Visualization of Strategic Workflows
Photoenzymatic Radical Reaction Development
Figure 1: A generalized workflow for developing a photoenzymatic radical reaction, highlighting the iterative process from target identification to successful implementation through enzyme engineering and condition optimization [3] [11].
Dual Photobiocatalysis Mechanism
Figure 2: Mechanism of the dual photobiocatalytic system for vicinal diamine synthesis, illustrating the synergy between an external photosensitizer (generating radicals) and an enzyme (controlling enantioselectivity) [4].
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Photoenzymatic Radical Synthesis
| Category | Reagent | Specific Function & Rationale |
|---|---|---|
| Enzyme Platforms | Ene-reductases (XenB, GluER, OYE1) | Catalyze new-to-nature radical reactions via photoexcited flavin; tunable via directed evolution [3] [5] [4] |
| Radical Precursors | Carbamates with N-O bonds (e.g., 4-cyanophenolate) | Generate N-centered radicals upon photoinduced electron transfer and N-O bond homolysis [3] |
| Fluorinated bromoamides (e.g., ethyl bromodifluoroacetamide) | Source of fluorinated carbon-centered radicals; amide group aids enzyme binding [5] | |
| N-amidopyridinium salts | Produce N-centered radicals via green-light-induced N-N bond cleavage [4] | |
| Cofactor Systems | NADP+/GDH/Glucose | Regenerates NADPH, which maintains flavin (FMN) in the reduced state (FMNH⁻) required for photoexcitation [3] [4] |
| Photosensitizers | Thioxanthone (genetically encoded) | Superior to benzophenone; absorbs visible light, highly efficient for energy transfer [11] |
| Rhodamine B (external) | Green-light-absorbing organic dye for synergistic catalysis with enzymes [4] | |
| Reaction Buffers | Imidazole Buffer (pH ~6.5) | Optimal for certain reactions like hydroamination; pH is a critical parameter [3] |
| Tris-HCl Buffer (pH ~8.5) | Alkaline buffer can enhance yield in other systems, such as fluorinated amide synthesis [5] |
Benchmarking Performance: Selectivity, Efficiency, and Green Metrics
The pursuit of sustainable and highly selective synthetic methodologies is a central goal in modern chemical research, particularly for the pharmaceutical industry. This application note provides a comparative analysis of two leading approaches: emerging photoenzymatic catalysis and established traditional chemocatalytic methods. Photoenzymatic catalysis merges the enantioselective power of enzymes with the unique reactivity unlocked by light, creating new-to-nature reactivities under mild conditions [45] [1]. In contrast, traditional chemocatalysis, including both homogeneous and heterogeneous catalysis, relies on the flexible reactivity of chemical catalysts, often requiring harsh temperatures and pressures [46] [47]. Framed within a broader thesis on enantioselective synthesis, this document provides a structured comparison of these paradigms, supported by quantitative data, detailed protocols for key photoenzymatic reactions, and essential resource lists to equip researchers in drug development.
Comparative Performance Analysis
The following tables summarize key performance metrics and characteristics of photoenzymatic and traditional chemocatalytic methods, based on recent literature.
Table 1: Quantitative Performance Comparison of Representative Reactions
| Reaction Type | Catalyst System | Yield (%) | Enantiomeric Excess (ee %) | Key Reaction Feature | Citation |
|---|---|---|---|---|---|
| Ketone Reduction | rGQDs/AKR (Photoenzymatic) | 82 | >99.99 | Cofactor-independent, H2O hydrogen source [36] | |
| Hydroalkylation of Alkenes | Ene-Reductase (Photoenzymatic) | Up to 91 | Up to 97 | Synthesis of fluorinated amides with distal chirality [5] | |
| C–H Functionalization | SD-VHbCH Carbene Transferase (Photoenzymatic) | Not Specified | Up to 99 | Enantioselective sp3 C–H functionalization [22] | |
| Hydrogenation | Nickel (Heterogeneous Chemocatalytic) | N/A | N/A | Requires high-pressure H2 gas, metal surface reaction [47] |
Table 2: Characteristic Comparison of Catalytic Methods
| Parameter | Photoenzymatic Catalysis | Traditional Chemocatalysis |
|---|---|---|
| Activation Energy | Lowered via enzymatic path and light energy [45] [47] | Lowered via alternative chemical mechanism [46] [47] |
| Typical Conditions | Mild, aqueous, room temperature [36] [5] | Often high temperature/pressure [45] |
| Sustainability Profile | High (Renewable catalysts, light energy, water solvent) [36] | Variable (Can involve precious metals, harsh chemicals) |
| Reaction Media | Often aqueous buffer, can tolerate co-solvents [5] | Organic solvents common [48] |
| Catalyst Recovery | Possible with engineered insoluble hybrids [36] | Straightforward for heterogeneous systems [46] |
Experimental Protocols
Protocol 1: Cofactor-Independent Photoenzymatic Ketone Reduction
This protocol describes the synthesis of (R)-1-[3,5-bis(trifluoromethyl)-phenyl] ethanol ((R)-3,5-BTPE), a pharmaceutical intermediate, using a hybrid photo-biocatalyst under infrared illumination [36].
- Workflow Diagram
Materials and Reagents
- Cross-linked Aldo-Keto Reductase (AKR-CLEs): The engineered enzyme scaffold [36].
- Reductive Graphene Quantum Dots (rGQDs): Infrared light-responsive nanomaterial [36].
- rGQDs/AKR Hybrid Photo-biocatalyst: Assembled from the above components [36].
- Ketone Substrate: 3,5-Bis(trifluoromethyl) acetophenone.
- Aqueous Buffer: Sodium phosphate or Tris-HCl buffer, pH ~7-8.5.
- IR Light Source: 980 nm laser or LED.
Step-by-Step Procedure
- Catalyst Preparation: Construct the rGQDs/AKR hybrid catalyst by grafting pre-synthesized rGQDs onto the cross-linked AKR (AKR-CLEs) through simple self-assembly in aqueous solution [36].
- Reaction Setup: Suspend the rGQDs/AKR catalyst and the prochiral ketone substrate in the appropriate aqueous buffer. Water serves as the ultimate hydrogen source [36].
- Illumination: Illuminate the reaction mixture with infrared light (e.g., 980 nm) with constant stirring. The rGQDs absorb IR light, mediating water splitting to generate active hydrogen atoms [36].
- Reaction Monitoring: Monitor reaction progress by analytical techniques such as GC or HPLC.
- Product Isolation & Catalyst Recovery: After completion, separate the insoluble rGQDs/AKR catalyst by centrifugation or filtration. Recover the chiral alcohol product, (R)-3,5-BTPE, from the supernatant by standard extraction and purification techniques. The hybrid catalyst can be recycled for subsequent runs [36].
Protocol 2: Photoenzymatic Enantioselective Hydroalkylation
This protocol details the synthesis of α-fluorinated amides with distal chirality via visible-light-driven ene-reductase catalysis [5].
- Workflow Diagram
Materials and Reagents
- Ene-Reductase (OYE1 mutant): Engineered flavin-dependent enzyme (e.g., Y375F mutant for enhanced yield) [5].
- Flavin Mononucleotide (FMN): Natural cofactor within the enzyme.
- Bromodifluoroacetamide Derivative (F-1): Radical precursor.
- Alkene: Electron-rich alkene coupling partner.
- Cofactor Recycling System: Glucose dehydrogenase (GDH), D-(+)-glucose, and NADP+.
- Buffer: Tris-HCl buffer, pH 8.5.
- Blue Light Source: 45 W blue LED (465 nm).
Step-by-Step Procedure
- Reaction Setup: In a suitable reaction vessel, combine the fluorinated reagent (F-1), alkene, OYE1 mutant (2 mol%), NADP+, GDH, and glucose in Tris-HCl buffer (pH 8.5). The GDH/glucose system ensures continuous regeneration of the NADPH required for the enzyme's catalytic cycle [5].
- Illumination: Seal the vessel and irradiate the mixture with vigorous stirring under blue light (465 nm) for the specified duration.
- Radical Process: Blue light excitation generates a carbon-centered radical from F-1. The ene-reductase then catalyzes the enantioselective addition of this fluorinated amide radical across the alkene [5].
- Reaction Quenching & Work-up: After completion, quench the reaction and extract the product using an organic solvent (e.g., ethyl acetate).
- Purification and Analysis: Purify the crude product by chromatography (e.g., flash silica gel). Analyze the yield and enantiomeric excess (ee) by techniques like GC-MS and chiral HPLC [5].
The Scientist's Toolkit
Table 3: Key Research Reagent Solutions for Photoenzymatic Catalysis
| Reagent / Material | Function in Photoenzymatic Catalysis | Example Application / Note |
|---|---|---|
| Reductive Graphene Quantum Dots (rGQDs) | IR-responsive photocatalyst; enables water splitting as hydrogen source [36]. | Cofactor-independent ketone reduction. Core component of hybrid catalyst. |
| Engineered Ene-Reductases (e.g., OYE1) | Flavoprotein catalyzing enantioselective radical additions under light [5]. | Hydroalkylation for fluorinated amide synthesis. Can be evolved for better performance. |
| Flavin Mononucleotide (FMN) | Natural photoactive cofactor; mediates single-electron transfer processes [5] [45]. | Essential for ene-reductase activity in radical reactions. |
| Carbene Transferase (e.g., SD-VHbCH) | Engineered enzyme for enantioselective carbene transfer from diazo compounds [22]. | Used in cascade C-H functionalization. |
| Cofactor Recycling System (GDH/Glucose) | Regenerates NADPH from NADP+ using glucose as sacrificial electron donor [5]. | Maintains catalytic turnover in NADPH-dependent enzymes. |
| Dual Nickel/Photocatalyst (Ni/PC) | Drives C-N cross-coupling in a photocatalytic step preceding biocatalysis [22]. | Used in multi-component cascade reactions. |
This comparative analysis delineates the distinct advantages and applications of photoenzymatic and traditional chemocatalytic methods. Photoenzymatic catalysis demonstrates unparalleled capabilities in achieving high enantioselectivity under mild, sustainable conditions, accessing radical intermediates, and enabling novel disconnections via cofactor-independent pathways [36] [5] [1]. Traditional chemocatalysis remains a robust and versatile tool for a wide range of transformations, particularly where enzymatic activity is incompatible. The choice between these methodologies depends on the specific synthetic challenge, with the emerging trend leaning toward hybrid systems that leverage the strengths of both worlds [48]. The provided protocols and toolkit aim to empower researchers in the selective adoption and implementation of these powerful catalytic strategies for advanced synthetic applications, including drug development.
Within the broader context of advancing photoenzymatic enantioselective synthesis, achieving high enantiomeric excess (ee) is a paramount objective, particularly for the production of pharmaceuticals and bioactive molecules where stereochemistry profoundly influences biological activity [5] [49]. This article presents application notes and detailed protocols for three exemplary photoenzymatic reactions that achieve >97% ee, a benchmark for high stereocontrol. These case studies underscore the power of combining visible light excitation with engineered enzymes to conduct challenging enantioselective transformations, such as radical hydrofunctionalizations, under mild and sustainable conditions [3] [4].
Case Studies in High-Efficiency Photoenzymatic Synthesis
The following table summarizes three recent breakthroughs in photoenzymatic synthesis that achieve exceptional enantioselectivity.
Table 1: Case Studies of Photoenzymatic Reactions with >97% Enantiomeric Excess
| Case Study | Reaction Type | Key Enzyme / Catalytic System | Maximum Reported ee (%) | Key Chiral Product |
|---|---|---|---|---|
| 1. Fluorinated Amide Synthesis [5] | Enantioselective hydroalkylation of alkenes | Ene-reductase (OYE1-Y375F mutant) | 97% | α-Fluorinated amides with distal chirality |
| 2. Intermolecular Radical Hydroamination [3] | Enantioselective intermolecular hydroamination of styrenes | Ene-reductase (XenB mutants, e.g., E1, E2) | 97% | Chiral amines (β-to-N benzylic stereocenter) |
| 3. Vicinal Diamine Synthesis [4] | Enantioselective hydroamination of enamides | Dual system: Ene-reductase (GluER-M5 mutant) & Rhodamine B (RhB) | >99% | Chiral vicinal diamines |
Experimental Protocols
This protocol describes the synthesis of enantiomerically enriched α-fluorinated amides via a visible-light-driven ene-reductase system.
Required Materials
- Enzyme: OYE1-Y375F mutant (2 mol%)
- Substrates: Fluorine-containing brominated amide (e.g., F-1), electron-rich alkene
- Cofactor Regeneration System: NADP+, Glucose Dehydrogenase (GDH), D-(+)-Glucose
- Solvent/Buffer: Tris-HCl buffer (pH 8.5)
- Light Source: 45 W blue LEDs (465 nm)
Step-by-Step Procedure
- Reaction Setup: In a glass vial, combine the fluorinated amide substrate (F-1, 1.0 equiv), the alkene (1.5 equiv), OYE1-Y375F mutant (2 mol%), NADP+ (catalytic amount), GDH (10 U), and D-glucose (5.0 equiv) in Tris-HCl buffer (pH 8.5).
- Photoirradiation: Seal the vial and place it under the irradiation of 45 W blue LEDs (465 nm). Maintain the reaction at room temperature with continuous stirring for 16-24 hours.
- Reaction Monitoring: Monitor the reaction progress by thin-layer chromatography (TLC) or GC-MS analysis.
- Work-up: After completion, extract the reaction mixture with ethyl acetate (3 × 10 mL). Combine the organic layers, dry over anhydrous Na₂SO₄, and concentrate under reduced pressure.
- Purification: Purify the crude product by flash chromatography on silica gel to obtain the desired α-fluorinated amide.
- Analysis: Determine enantiomeric excess (ee) by chiral HPLC or GC analysis.
This protocol outlines an enantioselective intermolecular hydroamination reaction catalyzed by an engineered ene-reductase, generating chiral amines with a remote stereocenter.
Required Materials
- Enzyme: XenB mutant (e.g., E1: W100L or E2: A232L, 1-2 mol%)
- Substrates: Carbamate radical precursor (e.g., 1a with 4-cyanophenolate leaving group), α-methyl styrene
- Cofactor Regeneration System: NADP+, Glucose Dehydrogenase (GDH), D-Glucose
- Solvent/Buffer: Imidazole buffer (pH 6.5)
- Light Source: Visible light source (e.g., blue LEDs)
Step-by-Step Procedure
- Reaction Setup: Charge a reaction vial with the carbamate substrate 1a (1.0 equiv), α-methyl styrene (2.0 equiv), the specified XenB mutant (1-2 mol%), NADP+ (catalytic amount), GDH, and D-glucose in imidazole buffer (pH 6.5).
- Photoirradiation: Place the reaction mixture under visible light irradiation (e.g., blue LEDs) at room temperature with stirring for 12-48 hours.
- Reaction Monitoring: Analyze aliquots via TLC or LC-MS to track consumption of the starting material.
- Work-up and Purification: Upon completion, extract the aqueous mixture with ethyl acetate, dry the combined organic extracts, and concentrate. Purify the residue by flash chromatography.
- Analysis: Determine the yield and enantiomeric excess (ee) of the product 3a using chiral analytical methods (e.g., chiral HPLC).
This protocol employs a synergistic photobiocatalytic system comprising an engineered ene-reductase and an organic dye for the enantioselective synthesis of vicinal diamines.
Required Materials
- Biocatalyst: GluER-M5 mutant (T36A-Y177F-F269V-K277M-Y343F, 1 mol%)
- Photocatalyst: Rhodamine B (RhB)
- Substrates: N-(1-phenylvinyl)-acetamide (1a), N-amidopyridinium salt (2a)
- Cofactor Regeneration System: NADP+, Glucose Dehydrogenase (GDH), D-Glucose
- Light Source: Green LEDs (530-540 nm)
Step-by-Step Procedure
- Reaction Setup: In a vial, combine the enamide substrate 1a (1.0 equiv), the N-amidopyridinium salt 2a (1.5 equiv), GluER-M5 mutant (1 mol%), Rhodamine B (2 mol%), NADP+, GDH, and D-glucose in an appropriate aqueous buffer.
- Photoirradiation: Irradiate the reaction mixture with green LEDs (530-540 nm) at room temperature with constant stirring for 24-48 hours.
- Reaction Monitoring: Monitor the reaction by LC-MS or TLC.
- Work-up and Purification: Extract the product with an organic solvent, dry the combined extracts, and concentrate. Purify the crude material via flash chromatography to isolate the vicinal diamine product (S)-3a.
- Analysis: Determine the chemical yield and enantiomeric excess (>99% ee) by chiral HPLC. The absolute configuration can be confirmed by single-crystal X-ray diffraction.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of these high-performance photoenzymatic protocols relies on key reagents and materials.
Table 2: Key Research Reagent Solutions for Photoenzymatic Synthesis
| Reagent / Material | Function & Application Notes |
|---|---|
| Engineered Ene-Reductases (EREDs) | Flavoenzymes repurposed to catalyze enantioselective radical reactions. Key to achieving high ee and reaction rate [5] [3]. |
| Flavin Mononucleotide (FMN) | Native cofactor of EREDs. Its reduced form (FMNH¯) acts as a potent single-electron donor upon photoexcitation [3]. |
| NADP+ / NADPH Cofactor System | Serves as the ultimate source of reducing equivalents. The system is regenerated in situ using GDH/glucose [5] [3] [4]. |
| N–O or N–N Bond Containing Precursors | Designed substrates (e.g., carbamates, N-amidopyridinium salts) that generate reactive N-centered radicals upon photoinduced single-electron transfer [3] [4]. |
| Organophotoredox Catalysts (e.g., Rhodamine B) | Externally added photocatalysts that work synergistically with enzymes to generate radicals under milder, biologically compatible light (e.g., green light) [4]. |
Workflow and Mechanism Visualization
The following diagram illustrates the general catalytic cycle for photoenzymatic radical hydrofunctionalization, integrating light absorption, electron transfer, and enantioselective bond formation.
General Photoenzymatic Radical Catalysis Cycle - This workflow shows the key stages from enzyme reduction to chiral product formation, highlighting the role of light-induced electron transfer.
The mechanism of enantiocontrol revolves around the enzyme's engineered active site, which pre-orients the prochiral substrate and the radical intermediate. The following diagram details this process for a generic alkene hydrofunctionalization.
Mechanism of Enantiocontrol in the Active Site - This sequence illustrates how the chiral enzyme environment dictates the face of radical addition and subsequent H-atom transfer to achieve high ee.
Analyzing Green Chemistry Credentials and Sustainability
The integration of green chemistry principles into synthetic methodology is fundamentally reshaping pharmaceutical and fine chemical research. This paradigm shift is particularly evident in the emerging field of photoenzymatic enantioselective synthesis, which combines the specificity of biocatalysis with the versatility of photochemistry. The 12 principles of green chemistry, first established by Paul Anastas and John Warner in 1998, provide a framework for designing chemical products and processes that reduce or eliminate the use and generation of hazardous substances [50]. These principles emphasize waste prevention, atom economy, reduced hazardous chemical use, and safer solvent systems—all critical considerations for sustainable drug development.
Within this context, photoenzymatic catalysis represents a transformative approach that addresses multiple green chemistry objectives simultaneously. By harnessing visible light as a traceless reagent and enzymes as highly selective catalysts, researchers can develop synthetic routes that bypass traditional energy-intensive and waste-generating processes. The pharmaceutical industry, in particular, stands to benefit substantially from these advancements, as pressure mounts to develop more sustainable manufacturing processes while maintaining the stereochemical precision required for bioactive molecules. This analysis examines the green chemistry credentials of contemporary photoenzymatic methods through the lens of specific case studies, quantitative sustainability metrics, and practical implementation protocols.
Green Chemistry Framework for Photoenzymatic Synthesis
Quantitative Sustainability Assessment Metrics
A comprehensive analysis of early-phase sustainability assessments for chemical processes reveals a diverse array of 53 distinct methods specifically suited for evaluating green chemistry technologies during initial development stages [51]. These assessment frameworks integrate traditional green chemistry metrics with broader sustainability indicators to provide multidimensional evaluation of emerging technologies. For photoenzymatic synthesis, several key metrics prove particularly relevant for establishing green credentials:
Table 1: Key Green Chemistry Metrics for Photoenzymatic Synthesis Evaluation
| Metric Category | Specific Metrics | Application to Photoenzymatic Synthesis |
|---|---|---|
| Material Efficiency | Atom Economy, Process Mass Intensity (PMI), E-factor | Measures efficient use of starting materials and minimization of waste in photobiocatalytic cascades |
| Energy Efficiency | Photon Efficiency, Reaction Mass Efficiency | Quantifies effective utilization of light energy and overall process efficiency |
| Environmental Impact | Life Cycle Assessment (LCA), Global Warming Potential | Evaluates reduced environmental footprint compared to conventional synthetic routes |
| Hazard Reduction | Safer Solvent Selection, Renewable Feedstocks | Assesses replacement of hazardous reagents with biocompatible aqueous systems |
The implementation of these metrics in early-stage research enables data-driven decision-making that aligns photoenzymatic method development with sustainability goals. For instance, the pharmaceutical industry has demonstrated the practical application of these principles through award-winning technologies, such as Merck's biocatalytic process for islatravir manufacturing, which replaced a 16-step chemical synthesis with a nine-enzyme cascade in a single aqueous stream [52]. This approach eliminated organic solvents, intermediate isolations, and workups, dramatically reducing process mass intensity while maintaining stereochemical precision—a guiding model for photoenzymatic method development.
Strategic Alignment with Green Chemistry Principles
Photoenzymatic synthesis directly addresses multiple principles of green chemistry through its fundamental operational mechanisms. The inherent atom economy of many photobiocatalytic transformations stems from their ability to construct complex molecular architectures from simple precursors in cascade reactions without requiring protecting groups or functional group interconversions. A representative example is the visible-light-driven ene-reductase system that generates fluorinated amides with high enantioselectivity (up to 97% ee) while achieving yields up to 91% [5]. This system exemplifies principle #2 (atom economy) by efficiently incorporating starting materials into the final product while minimizing wasteful protection/deprotection sequences.
The energy efficiency of photoenzymatic methods (principle #6) derives from the use of visible light as an abundant, renewable energy source that operates under mild reaction conditions, significantly reducing the energy requirements associated with traditional thermal activation. Recent advances in enzyme engineering have further enhanced this aspect, with thioxanthone-based photoenzymes demonstrating remarkable efficiency (kcat = 13 s⁻¹, >1,300 turnovers) under visible light irradiation [11]. This represents a substantial improvement over conventional UV-absorbing systems and reduces the environmental footprint associated with specialized lighting infrastructure.
The solvent selection in photoenzymatic systems (principle #5) typically employs aqueous buffers or biocompatible solvent mixtures, eliminating the need for hazardous organic solvents commonly used in traditional synthesis. The inherent catalytic efficiency (principle #9) of these systems combines the exceptional turnover of photocatalysts with the exquisite selectivity of enzymes, minimizing stoichiometric waste and enabling operation at low catalyst loadings. Furthermore, the biodegradability of the biocatalytic components (principle #10) reduces environmental persistence concerns associated with transition metal catalysts that dominate conventional asymmetric synthesis.
Case Study: Photoenzymatic Synthesis of Fluorinated Amides
Experimental Protocol and Workflow
Protocol: Photoenzymatic Enantioselective Synthesis of α-Fluorinated Amides via Ene-Reductases
Reaction Setup: In a 5 mL glass vial equipped with a magnetic stir bar, combine the fluorinated bromoamide precursor (F-1, 0.2 mmol), electron-deficient alkene (0.24 mmol), glucose (2.0 mmol), NADP⁺ (0.02 mmol), and glucose dehydrogenase (GDH, 5 mg) in Tris-HCl buffer (2.0 mL, 100 mM, pH 8.5). Add the ene-reductase enzyme (2 mol%, typically OYE1 or engineered variant) to the reaction mixture [5].
Photoreaction Conditions: Seal the vial with a septum and place it 5 cm from a blue LED array (465 nm, 45 W). Irradiate the reaction mixture while stirring at 25°C for 24 hours. Monitor reaction progress by regular sampling and GC-MS or HPLC analysis.
Workup and Isolation: After completion, extract the reaction mixture with ethyl acetate (3 × 5 mL). Combine the organic layers, dry over anhydrous MgSO₄, filter, and concentrate under reduced pressure. Purify the crude product by flash chromatography on silica gel to obtain the desired fluorinated amide.
Analytical Characterization: Determine enantiomeric purity by chiral HPLC or SFC analysis. Confirm structural identity by NMR spectroscopy and high-resolution mass spectrometry.
Key Optimization Parameters:
- Light Intensity: Systematic variation between 12W and 45W demonstrated improved conversion at higher power [5]
- Buffer System: Alkaline conditions (Tris-HCl, pH 8.5) enhanced yield by 1.7-fold compared to phosphate buffers [5]
- Enzyme Loading: Increasing to 2 mol% provided optimal balance between activity and practical implementation
Green Chemistry Performance Assessment
This photoenzymatic fluorination methodology demonstrates substantial advantages over conventional approaches through multiple green chemistry metrics:
Table 2: Comparative Analysis of Fluorinated Amide Synthesis Methods
| Parameter | Traditional Transition Metal Catalysis | Photoenzymatic Approach | Green Chemistry Benefit |
|---|---|---|---|
| Catalyst System | Palladium/nickel complexes | Ene-reductases + visible light | Replaces precious metals with renewable biocatalysts |
| Solvent System | Anhydrous organic solvents (DMF, THF) | Aqueous buffer | Eliminates hazardous solvent use and generation |
| Reaction Conditions | Elevated temperatures, inert atmosphere | Ambient temperature, aerobic | Reduces energy consumption and specialized equipment |
| Stereocontrol | Requires chiral ligands/auxiliaries | Intrinsic enzyme enantioselectivity | Eliminates stoichiometric chiral directing groups |
| Waste Generation | Metal residues, ligand decomposition | Biodegradable enzyme proteins | Reduces hazardous waste streams |
The atom-economic profile of this transformation is particularly favorable, as it directly incorporates the fluorinated moiety without requiring functional group manipulation. The system's ability to generate fluorinated amides with distal chirality (γ-to F) with high enantiocontrol (up to 97% ee) addresses a significant challenge in conventional asymmetric synthesis, where installation of stereocenters remote to functional groups typically requires multi-step sequences with accumulating material inefficiencies [5].
Advanced Photoenzymatic Systems and Engineering Strategies
Artificial Photoenzyme Design and Implementation
Protocol: Development of Genetically Encoded Thioxanthone Photoenzymes
Non-Canonical Amino Acid Synthesis: Prepare thioxanthon-2-ylalanine (mTX) or thioxanthon-3-ylalanine (pTX) according to published procedures [11]. These amino acids serve as genetically encodable triplet sensitizers with superior photophysical properties compared to natural aromatic amino acids.
tRNA Synthetase Engineering: Evolve orthogonal Methanococcus jannaschii tyrosyl-tRNA synthetase/tRNA pairs for specific incorporation of mTX or pTX into protein scaffolds. Employ a GFP-based selection system to identify variants with high activity and specificity [11].
Photoenzyme Assembly: Incorporate mTX into predetermined positions within protein scaffolds (e.g., DA2000 at position 173) via genetic code expansion. Screen library variants for photocatalytic activity and enantioselectivity in model [2+2] cycloadditions.
Directed Evolution: Randomize 24-30 individual positions using NNK degenerate codons. Assay approximately 5,300 clones under 405 nm irradiation to identify mutants with enhanced activity and selectivity [11]. Iterate through multiple rounds (typically 3-4) to optimize performance.
The resulting thioxanthone-based photoenzymes demonstrate remarkable improvements over earlier benzophenone-containing systems, with a 10-fold increase in reaction yield and the ability to achieve >1,300 turnovers [11]. This enhanced efficiency directly translates to improved sustainability metrics through reduced catalyst loading and increased productivity.
Integrated Photoenzymatic Cascade Systems
Protocol: One-Pot Photo-Enzymatic Cascade Multi-Component Reaction
Reaction Design: Combine nickel/photoredox dual catalysis with engineered carbene transferases in a single reaction vessel to achieve enantioselective C(sp³)–H functionalization of saturated N-heterocyclic scaffolds [22].
Photocatalytic Step: In a dried glass reactor, combine aryl bromide (0.1 mmol), cyclic secondary amine (0.12 mmol), Ni(COD)₂ (5 mol%), photocatalyst (3 mol%), and DMSO (1 mL). Irradiate with blue LEDs (450 nm) under nitrogen atmosphere for 6-12 hours to generate saturated N-heterocyclic intermediates in situ.
Biocatalytic Functionalization: Without intermediate purification, add the whole-cell system containing engineered SD-VHbCH carbene transferase (20 mg), sodium dithionite (10 mM), and donor reagent (0.15 mmol) directly to the reaction mixture. Incubate at 30°C with shaking for 24 hours to obtain chiral α-functionalized phenylpyrrolidine derivatives.
Process Optimization: Utilize computational studies to identify critical active pocket residues influencing stereoselectivity. Engineer binding pockets to provide more stable reaction environments that enhance enantiocontrol (up to 99% ee) [22].
This cascade methodology exemplifies the circular design principles of green chemistry by eliminating intermediate isolation and purification steps, significantly reducing solvent consumption and energy input compared to sequential synthetic approaches. The ability to functionalize inert C-H bonds with high stereocontrol represents a fundamental advancement in synthetic efficiency, bypassing the need for pre-functionalized substrates and the associated waste generation.
Diagram 1: Artificial Photoenzyme Engineering Workflow. This flowchart illustrates the key stages in developing genetically encoded photoenzymes, from initial design to application.
Research Reagent Solutions for Photoenzymatic Synthesis
Table 3: Essential Research Reagents for Photoenzymatic Method Development
| Reagent Category | Specific Examples | Function and Application | Sustainability Considerations |
|---|---|---|---|
| Ene-Reductases | OYE1, OYE2, OYE3, XenA, XenB | Catalyze asymmetric reduction and radical hydroalkylation of alkenes | Renewable biocatalysts replace precious metal catalysts |
| Flavin-Dependent Enzymes | Flavoprotein photoreceptors | Enable net-reductive and redox-neutral photoenzymatic catalysis | Utilize naturally occurring cofactors (FMN, FAD) |
| Non-Canonical Amino Acids | Thioxanthon-2-ylalanine (mTX), Thioxanthon-3-ylalanine (pTX) | Serve as genetically encodable triplet sensitizers in artificial photoenzymes | Enable visible light absorption; reduce UV-associated hazards |
| Natural Photoenzymes | DNA photolyases, BLUF domains | Provide native protein scaffolds for repurposing to new transformations | Demonstrate natural precedent for photobiocatalytic mechanisms |
| Cofactor Recycling Systems | Glucose/GDH, formate/FDH | Regenerate reduced nicotinamide cofactors (NAD(P)H) | Minimize stoichiometric cofactor use; improve atom economy |
| Engineered Carbene Transferases | SD-VHbCH variants | Catalyze stereoselective carbene insertion reactions in cascade systems | Enable C-H functionalization without directing groups |
The selection of appropriate reagent systems is critical for optimizing both the synthetic efficiency and sustainability profile of photoenzymatic methodologies. The trend toward genetically encoded photosensitizers represents a particularly promising direction, as it eliminates the need for external photosensitizers that can complicate purification and contribute to waste streams. Similarly, the development of aerobic-tolerant photoenzymes addresses a significant practical limitation of many photoredox systems that require strict oxygen exclusion, reducing energy consumption associated with degassing procedures [11].
The systematic analysis of green chemistry credentials in photoenzymatic enantioselective synthesis reveals a technology platform with inherent sustainability advantages across multiple dimensions. The quantitative metrics presented in this analysis demonstrate tangible improvements in atom economy, energy efficiency, and waste reduction compared to conventional synthetic approaches. These advancements align with the growing emphasis on early-phase sustainability assessments in chemical process development, as evidenced by the emergence of specialized evaluation frameworks for nascent technologies [51].
Future developments in photoenzymatic synthesis will likely focus on addressing remaining challenges in scalability, cofactor dependency, and reaction scope. The integration of artificial intelligence and machine learning approaches for enzyme design and reaction optimization represents a particularly promising direction, potentially accelerating the development of next-generation photoenzymes with enhanced catalytic properties and broader substrate scope [50]. Additionally, the continued expansion of genetic code expansion methodologies will enable incorporation of increasingly sophisticated photocatalytic moieties into protein scaffolds, further blurring the boundaries between natural enzyme function and synthetic photocatalysis.
As pharmaceutical and fine chemical industries face increasing pressure to adopt more sustainable manufacturing practices, photoenzymatic enantioselective synthesis offers a compelling technological pathway that aligns synthetic efficiency with environmental responsibility. By embedding green chemistry principles into fundamental reaction design from the earliest research phases, the field is poised to deliver transformative synthetic methodologies that simultaneously advance molecular innovation and sustainability objectives.
Computational Validation of Mechanisms and Selectivity Origins
Photoenzymatic catalysis represents a rapidly advancing frontier in synthetic chemistry, merging the remarkable stereocontrol of enzymes with the versatile activation modes of photochemistry. A critical challenge in this field lies in moving beyond empirical discovery to gain a precise, mechanistic understanding of how these biocatalysts control reactivity and selectivity, particularly for non-natural radical transformations. Computational methods have emerged as indispensable tools for validating proposed mechanisms and elucidating the atomic-level origins of enantioselectivity. This Application Note details protocols for computational investigation of photoenzymatic systems, drawing from recent advances in the enantioselective synthesis of fluorinated compounds, amines, and other high-value chiral building blocks. Framed within a broader thesis on photoenzymatic enantioselective synthesis, this guide provides researchers with methodologies to computationally validate radical mechanisms and rationalize stereocontrol, thereby accelerating the design of more efficient and selective biocatalysts.
Key Experimental Data and Computational Insights
Recent landmark studies have successfully combined photoenzymatic synthesis with computational validation. The quantitative data and key insights from these studies are summarized in the table below for direct comparison.
Table 1: Computational Insights from Recent Photoenzymatic Studies
| Reaction Type | Key Computational Findings | Impact on Yield/Selectivity | Primary Computational Methods |
|---|---|---|---|
| Fluorinated Amide Synthesis [5] | Amide substrates showed stronger enzyme binding than esters; Y375F mutation enhanced radical binding affinity. | Yield increased to >90%; enantiomeric excess (e.e.) maintained at >90%. | Molecular Docking, Binding Energy Calculations |
| Radical Hydroamination [3] | Enantioselectivity originates from the radical-addition step, not the H-atom transfer step. | Achieved up to 97% e.e. in intermolecular radical hydroamination. | Mechanistic Analysis, Reaction Pathway Modeling |
| Red Light Photoenzyme Engineering [53] | Cyan absorption: π→π* on flavin. Red absorption: π→π* between flavin and substrate, enabled by altered binding conformation. | Enabled reaction scale-up to 10 g using lower-energy red light. | MD Simulations, Docking, Excited-State Calculations |
| Spirocyclic β-Lactam Synthesis [11] | Engineered enzyme active site suppresses competing substrate decomposition pathway observed with small-molecule sensitizers. | Achieved 99% e.e. and 22:1 d.r.; enhanced reaction efficiency and selectivity. | Protein Design, Transition State Modeling |
| One-Pot Hydroxysulfone Synthesis [23] | MD simulations revealed enzyme-substrate interactions responsible for activity and stereoselectivity; S96 and Y190 mutations opened binding pocket. | Yield reached 99% with 99% e.e. | Molecular Dynamics (MD), Docking |
Computational Methodologies and Protocols
Protocol 1: Molecular Docking and Binding Affinity Analysis
This protocol is essential for rationalizing substrate specificity and guiding enzyme engineering, as demonstrated in the development of ene-reductases for fluorinated amide synthesis [5].
- Step 1: System Preparation
- Enzyme Structure: Obtain a crystal structure of the target enzyme (e.g., OYE1) from the PDB. Use protein preparation tools (e.g., in Maestro or MOE) to add hydrogens, assign protonation states, and optimize hydrogen bonding networks.
- Ligand Structure: Generate 3D structures of the substrate and radical intermediate using chemical drawing software (e.g., ChemDraw). Perform geometry optimization with semi-empirical or DFT methods (e.g., PM6, B3LYP/6-31G*).
- Step 2: Molecular Docking
- Define the active site as a grid box centered on the native ligand or key catalytic residues.
- Use docking software (e.g., AutoDock Vina or GOLD) to generate multiple binding poses. Set the number of poses to 20 and the exhaustiveness to 16 (for Vina) for a comprehensive search.
- Step 3: Pose Analysis and Binding Energy Calculation
- Analyze the top-ranked poses for key interactions (e.g., π-stacking, H-bonding, van der Waals contacts).
- Calculate relative binding energies to compare different substrates or mutant enzymes, as was key to understanding the preference for amide over ester substrates [5].
Protocol 2: Molecular Dynamics (MD) for Stereoselectivity Analysis
MD simulations provide dynamic insight into how enzyme mutations allosterically tune active site properties, crucial for explaining phenomena like red-shifted absorption in engineered photoenzymes [53].
- Step 1: System Setup
- Build the simulation system using the docked enzyme-ligand complex. Solvate the protein in a cubic TIP3P water box with a 10 Å buffer. Add ions (e.g., Na+, Cl-) to neutralize the system and achieve a physiological salt concentration of 0.15 M.
- Step 2: Simulation Run
- Perform energy minimization (5,000 steps), followed by equilibration in the NVT (100 ps) and NPT (100 ps) ensembles.
- Run a production MD simulation for a minimum of 100 ns. Use a 2 fs integration time step and maintain temperature and pressure at 300 K and 1 bar using a Langevin thermostat and Berendsen barostat.
- Step 3: Trajectory Analysis
- Calculate the root-mean-square deviation (RMSD) to assess system stability.
- Analyze root-mean-square fluctuation (RMSF) of residues to identify flexible regions.
- Use the MM-GBSA method to calculate binding free energies from simulation snapshots. Cluster analysis of ligand poses can reveal the dominant binding modes responsible for stereocontrol.
Protocol 3: Excited-State Calculations for Spectral Tuning
This protocol is used to model enzyme-templated charge-transfer (CT) complexes and understand how the protein environment influences their photophysical properties [53].
- Step 1: Model System Construction
- Extract representative snapshots of the CT complex (flavin + substrate) from the MD trajectory.
- Create a truncated quantum mechanics/molecular mechanics (QM/MM) model, treating the flavin and substrate with QM and the surrounding protein with MM.
- Step 2: Electronic Structure Calculation
- Optimize the ground-state geometry of the QM region using DFT (e.g., ωB97X-D/6-31G*).
- Perform time-dependent DFT (TD-DFT) calculations on the optimized structure to compute vertical excitation energies and simulate the UV-visible absorption spectrum.
- Step 3: Spectral Assignment
- Analyze the molecular orbitals involved in the electronic transitions. As demonstrated [53], this can distinguish a π→π* transition localized on the flavin (cyan absorption) from a π→π* transition between flavin and substrate (red absorption).
- Correlate spectral shifts with specific mutations or changes in the binding conformation.
The following diagram illustrates the logical workflow integrating these computational methods to validate mechanisms and selectivity in photoenzymatic catalysis.
The Scientist's Toolkit: Essential Research Reagents and Materials
The table below lists key reagents and materials used in the featured computational and experimental studies.
Table 2: Key Research Reagent Solutions for Photoenzymatic Studies
| Reagent/Material | Function/Application | Example from Literature |
|---|---|---|
| Ene-Reductases (EREDs) | Flavin-dependent enzymes catalyzing enantioselective radical reactions via photoinduced electron transfer. | OYE1, XenB, GluER used for hydroalkylation, hydroamination, and cyclization [5] [3] [53]. |
| Engineered Ketoreductases (KREDs) | NAD(P)H-dependent enzymes for asymmetric reduction of prochiral ketones to chiral alcohols. | Engineered LkKRED for synthesis of chiral hydroxysulfones with 99% ee [23]. |
| Non-Canonical Amino Acids | Genetically encoded sensitizers (e.g., thioxanthone) to expand enzyme photophysical properties. | mTX for visible-light-driven [2+2] cycloadditions [11]. |
| Flavin Mononucleotide (FMN) | Native cofactor in EREDs; its reduced form (FMNH-) acts as a potent photoexcited reductant. | Key for generating carbon- and nitrogen-centered radicals [3]. |
| NADP+/Glucose Dehydrogenase | Cofactor regeneration system; sustains catalytic cycles by recycling NADPH. | Used in photoenzymatic hydroamination and hydroalkylation systems [5] [3]. |
| Molecular Docking Software | Predicts substrate orientation and binding affinity within the enzyme active site. | AutoDock Vina used to identify key residues for engineering [5] [23]. |
| MD Simulation Packages | Models the dynamic behavior of enzyme-ligand complexes in a solvated environment. | GROMACS/AMBER for analyzing conformational changes and allostery [53]. |
| Quantum Chemistry Codes | Performs excited-state calculations to model charge-transfer complexes and spectral properties. | Used for TD-DFT calculations on flavin-substrate complexes [53]. |
Computational methods have transitioned from supportive roles to central tools for validating mechanisms and decoding selectivity in photoenzymatic synthesis. The synergistic combination of molecular docking, MD simulations, and excited-state calculations provides a powerful framework for understanding how enzyme active sites control the fate of highly reactive radical intermediates. This understanding, as detailed in the provided protocols, enables the rational engineering of photoenzymes for enhanced activity, altered selectivity, and even new reactivity, as evidenced by the development of systems operating with red light. As these computational strategies continue to evolve, they will undoubtedly accelerate the discovery and optimization of next-generation photoenzymes, expanding the toolbox for sustainable asymmetric synthesis in pharmaceutical and fine chemical development.
Practical Scalability and Applications in Pharmaceutical Synthesis
Photoenzymatic catalysis represents an emerging paradigm in synthetic chemistry that integrates the remarkable stereocontrol of enzymatic catalysis with the versatile reactivity of light-driven processes. This synergistic approach has established itself as a pivotal tool for asymmetric synthesis, enabling access to valuable chiral building blocks that are notoriously difficult to produce using traditional methods [2]. The field has progressed significantly through the repurposing of natural enzymes, particularly flavin-dependent 'ene'-reductases (EREDs), to catalyze diverse unnatural transformations under visible light irradiation [2] [19]. These biocatalytic systems offer exquisite control over chemo-, enantio-, and substrate selectivity while operating under mild, environmentally benign conditions [19]. The practical implementation of photoenzymatic methodologies in pharmaceutical synthesis addresses growing industry demands for sustainable manufacturing processes that reduce waste generation and energy consumption [54]. This application note examines recent advances in photoenzymatic synthesis with emphasis on scalability, experimental protocols, and implementation in drug development pipelines.
The tables below summarize performance data for key photoenzymatic transformations relevant to pharmaceutical synthesis, demonstrating the efficiency and selectivity achievable with these methods.
Table 1: Performance Metrics for Photoenzymatic Synthesis of Fluorinated Compounds
| Reaction Type | Yield Range (%) | Enantiomeric Excess (ee%) | Key Enzyme | Reference |
|---|---|---|---|---|
| Fluorinated Amide Synthesis | up to 91% | up to 97% | Engineered OYE1 | [5] |
| Hydroalkylation with Fluorinated Amides | 51-91% | 90-97% | OYE1-Y375F Mutant | [5] |
| Radical Hydroamination | 25-96% | 88-97% | Evolved XenB Mutants | [3] |
| Oxygen-Containing Heterocycle Synthesis | High yields | Great stereoselectivities | GluER-W100H | [55] |
Table 2: Scalability Assessment of Photoenzymatic Platforms
| Reaction System | Gram-scale Demonstrated | Temperature | Light Source | Cofactor Recycling |
|---|---|---|---|---|
| Ene-Reductase System | Yes | Room Temperature | Blue LED (455-465 nm) | GDH/Glucose/NADP+ [5] |
| XenB Hydroamination | Yes | Room Temperature | Visible Light | GDH/Glucose/NADP+ [3] |
| Photoenzymatic Cascade | Not specified | Room Temperature | Blue LED | Integrated [22] |
Experimental Protocols
Photoenzymatic Synthesis of Fluorinated Amides
Background: This protocol describes the enantioselective synthesis of α-fluorinated amides with distal chirality (γ-to F) using a visible-light-driven ene-reductase system [5]. The method addresses the significant challenge of enantioselective synthesis of fluorinated amides, which are prevalent structural motifs in pharmaceuticals and bioactive molecules.
Materials:
- Ene-reductase (OYE1 wild-type or Y375F mutant)
- Fluorine-containing brominated amide substrate (F-1)
- Electron-rich alkene coupling partner
- Flavin mononucleotide (FMN) cofactor
- Glucose dehydrogenase (GDH)
- D-(+)-glucose
- NADP+
- Tris-HCl buffer (pH 8.5)
- Anaerobic reaction vessel with quartz window
- Blue LED light source (45 W, 465 nm)
Procedure:
- Prepare reaction mixture in Tris-HCl buffer (pH 8.5) containing NADP+ (0.5 mM), FMN (0.1 mM), and GDH (1 mg/mL).
- Add D-(+)-glucose (50 mM) as terminal electron donor for cofactor regeneration.
- Incorporate ene-reductase (2 mol% relative to substrate) and substrates (0.1-1.0 M concentration range).
- Transfer reaction mixture to anaerobic vessel and degas with inert gas (N₂ or Ar) for 10 minutes.
- Illuminate with blue LED light source (45 W, 465 nm) with continuous stirring for 24-48 hours.
- Monitor reaction progress by TLC, GC-MS, or HPLC.
- Terminate reaction by extracting with ethyl acetate (3 × 20 mL).
- Purify combined organic layers by flash chromatography to isolate products.
- Characterize products by NMR, HRMS, and determine enantiomeric excess by chiral HPLC.
Technical Notes:
- The Y375F mutation enhances yield (>90%) while maintaining stereoselectivity (>90% ee) [5].
- Alkaline buffer (Tris-HCl pH 8.5) significantly improves reaction efficiency compared to neutral buffers.
- Substrate concentration can be optimized based on solubility and enzyme tolerance.
- Control experiments confirm necessity of both enzyme and light for product formation.
Photoenzymatic Intermolecular Radical Hydroamination
Background: This protocol describes a pure biocatalytic system for enantioselective intermolecular radical hydroamination, repurposing an ene-reductase through directed evolution [3]. The method addresses the long-standing challenge of catalytic enantioselective intermolecular radical hydroamination.
Materials:
- Evolved XenB variants (W100L or A232L mutants)
- Carbamate substrate (protected primary aliphatic amine with N-O bond)
- α-methyl styrene or derivative
- Imidazole buffer (pH 6.5)
- NADP+, GDH, and glucose for cofactor regeneration
- Visible light source
- Anaerobic reaction setup
Procedure:
- Prepare reaction mixture in imidazole buffer (pH 6.5) containing NADP+ (0.5 mM), GDH (1 mg/mL), and glucose (50 mM).
- Add evolved XenB variant (2 mol%) and substrates (carbamate and olefin in 1:1.5 ratio).
- Transfer to anaerobic vessel and degas with inert gas.
- Illuminate with visible light (standard conditions) or shorter wavelengths for enhanced reactivity.
- Monitor reaction over 24-48 hours.
- Extract with ethyl acetate (3 × 20 mL) and combine organic layers.
- Purify by flash chromatography to isolate hydroamination products.
- Characterize products and determine enantiomeric excess (typically 93-97% ee) by chiral HPLC.
Technical Notes:
- pH and buffer type are critical factors influencing reaction outcome [3].
- Optimal yields observed under slightly acidic conditions (imidazole buffer, pH = 6.5).
- Directed evolution generated mutants with improved reactivity and enantioselectivity.
- Scalability demonstrated with gram-scale reactions maintaining efficiency and selectivity.
Enzyme Engineering and Directed Evolution Protocol
Background: This protocol describes the directed evolution approach for improving photoenzyme performance, based on successful engineering campaigns for ene-reductases [5] [3].
Materials:
- Wild-type enzyme (OYE1, XenB, or homolog)
- Site-directed mutagenesis kit
- High-throughput screening platform
- Expression system (E. coli or similar)
- Analytical equipment (HPLC, GC-MS)
Procedure:
- Library Design: Identify target residues through structural analysis and computational docking. Focus on residues near substrate binding pocket (e.g., Y375, Y196, F123, W116 in OYE1) [5].
- Mutagenesis: Create focused mutant library using site-saturation mutagenesis at identified positions.
- Expression: Express variant libraries in suitable host system (E. coli).
- Screening: Implement high-throughput screening for reactivity and enantioselectivity under photo reaction conditions.
- Hit Validation: Confirm improved variants in small-scale reactions with detailed analysis.
- Iteration: Perform additional rounds of mutagenesis and screening combining beneficial mutations.
Technical Notes:
- Key mutations (Y375F in OYE1, W100L/A232L in XenB) significantly enhanced both yield and enantioselectivity [5] [3].
- Computational studies and molecular docking guide rational library design.
- Balance between library size and quality is essential for efficient evolution campaigns.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Photoenzymatic Synthesis
| Reagent/Catalyst | Function | Application Notes |
|---|---|---|
| Ene-Reductases (OYE1, XenB) | Flavin-dependent biocatalysts for radical generation & chiral control | Engineered via directed evolution; tolerate diverse substrates [5] [3] |
| Flavin Mononucleotide (FMN) | Photoredox cofactor for electron transfer | Generates carbon-/nitrogen-centered radicals upon light excitation [19] [3] |
| Glucose Dehydrogenase (GDH) | Cofactor regeneration system | Maintains NADPH pool for sustained catalysis [5] [3] |
| NADP+ | Redox cofactor | Electron shuttle between GDH and photoenzyme |
| Fluorinated Amide Precursors | Radical precursors for fluorinated compounds | Designed with leaving groups for efficient radical generation [5] |
| Carbamate Substrates | Nitrogen-centered radical precursors | Feature N-O bonds for homolytic cleavage upon reduction [3] |
Workflow and Mechanism Visualization
Diagram 1: Photoenzymatic Catalysis Cycle illustrating the integrated light activation, radical generation, and asymmetric bond formation steps with cofactor regeneration.
Diagram 2: Implementation Workflow for integrating photoenzymatic synthesis in pharmaceutical development, highlighting key optimization parameters.
Applications in Pharmaceutical Synthesis
Photoenzymatic methods have significant implications for pharmaceutical synthesis, particularly in constructing challenging molecular architectures with biological relevance. The synthesis of fluorinated amides is especially valuable given that fluorine-containing drugs now surpass 50% of all pharmaceuticals [5]. The incorporation of gem-difluoro/monofluoro groups alters physical, chemical, and biological properties of drug candidates due to fluorine's favorable biocompatibility, high electronegativity, and metabolic stability [5]. Similarly, chiral amine synthesis via radical hydroamination provides access to key building blocks for bioactive molecules, natural products, and resolution agents with applications across pharmaceutical, agrochemical, and material sectors [3].
The scalability of these photoenzymatic systems has been demonstrated through gram-scale reactions that maintain efficiency and selectivity [3]. The mild reaction conditions (room temperature, aqueous buffers, visible light irradiation) and eliminated requirement for precious metal catalysts contribute to more sustainable manufacturing processes aligned with green chemistry principles [54]. The enzyme engineering platforms developed for these systems enable optimization of activity, selectivity, and stability for specific industrial applications, facilitating implementation in diverse synthetic routes [5] [3] [54].
Photoenzymatic enantioselective synthesis represents a rapidly advancing field with demonstrated practical applications in pharmaceutical synthesis. The protocols and data presented herein provide researchers with actionable methodologies for implementing these techniques in drug development programs. The continued evolution of photoenzyme engineering, reactor design, and process optimization will further enhance the scalability and industrial adoption of these sustainable biocatalytic platforms. As the field progresses, integration of artificial intelligence and automated screening platforms promises to accelerate the development of next-generation photoenzymes with tailored selectivity profiles for specific synthetic challenges [56].
Conclusion
Photoenzymatic enantioselective synthesis represents a paradigm shift in asymmetric catalysis, successfully addressing long-standing challenges in controlling radical intermediates with exceptional stereoselectivity. By integrating the foundational principles of photochemistry with the evolvability of enzymes, this approach enables efficient access to structurally diverse and pharmaceutically relevant chiral molecules, including fluorinated amides, amines, and diamines, that are difficult to obtain by conventional means. The future of this field lies in the continued engineering of enzyme platforms for broader substrate scope and novel reactivities, the deeper mechanistic understanding of selectivity through advanced computational models, and the seamless integration of these methods into the synthesis of complex active pharmaceutical ingredients. For biomedical research, these advancements promise to accelerate the discovery and development of new therapeutic agents with optimized properties, underscoring the transformative potential of photoenzymatic catalysis in clinical research and sustainable drug manufacturing.
References
- 1. Bridging chemistry and biology for light-driven new-to- ... [https://pubs.rsc.org/en/content/articlelanding/2025/cs/d4cs00561a]
- 2. Recent advances in repurposing natural enzymes for new- ... [https://pubs.rsc.org/en/content/articlehtml/2025/qo/d5qo00470e]
- 3. Photoenzymatic Enantioselective Intermolecular Radical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10948044/]
- 4. Enantioselective biosynthesis of vicinal diamines enabled ... [https://www.sciencedirect.com/science/article/abs/pii/S1872206724601683]
- 5. Photoenzymatic enantioselective synthesis of fluorinated ... [https://www.nature.com/articles/s41467-025-60807-0]
- 6. Threonine Aldolase-Catalyzed Enantioselective α ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11376206/]
- 7. Thermal, electrochemical and photochemical reactions ... [https://www.sciencedirect.com/science/article/abs/pii/S1874604720300159]
- 8. A New Thermophilic Ene-Reductase from the Filamentous ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8146883/]
- 9. Reductases Catalyzed Non-Natural Radical Reactions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11618812/]
- 10. Flavoprotein Photochemistry: Fundamental Processes and ... [https://pubmed.ncbi.nlm.nih.gov/35442696/]
- 11. Efficient and selective energy transfer photoenzymes ... [https://www.nature.com/articles/s41557-025-01820-0]
- 12. intermolecular radical... | Nature Photoenzymatic enantioselective [https://www.nature.com/articles/s41586-020-2406-6?error=cookies_not_supported&code=a202bade-626a-4c57-bc26-973d18316160]
- 13. Steering ene-reductases for non-natural C(sp2)-C(sp3) couplings... [https://communities.springernature.com/posts/steering-ene-reductases-for-non-natural-c-sp2-c-sp3-couplings-through-photoinduced-single-electron-oxidation]
- 14. biosynthesis of vicinal diamines enabled by... Enantioselective [https://www.cjcatal.com/EN/abstract/abstract30237.shtml]
- 15. Radical Hydrocyanoalkylation of Alkenes via... Enantioselective [https://pubmed.ncbi.nlm.nih.gov/40747066/]
- 16. Photo-excited extracellular electron transfer of electroactive ... [https://www.nature.com/articles/s41467-025-65119-x]
- 17. Charge-transfer complex [https://en.wikipedia.org/wiki/Charge-transfer_complex]
- 18. EDA complex photochemistry as a strategy for C–S bond ... [https://pubs.rsc.org/en/content/articlehtml/2025/qo/d5qo00258c]
- 19. Molecular Origins of Simultaneous Chemo‑, Enantio‑, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12616699/]
- 20. Dioxygen compatible electron donor-acceptor catalytic ... [https://www.nature.com/articles/s41467-024-45866-z]
- 21. Non-Aufbau electronic structure in radical enzymes and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11290419/]
- 22. Enantioselective synthesis of α-functionalized ... [https://pubs.rsc.org/en/content/articlelanding/2025/qo/d5qo00169b]
- 23. Photo-Biocatalytic One-Pot Cascade Reaction for the ... [https://www.mdpi.com/2073-4344/15/8/733]
- 24. Illinois Data Bank [https://databank.illinois.edu/datasets/IDB-7913244]
- 25. Ru(II) photocages enable precise control over enzyme ... [https://www.nature.com/articles/s41467-022-31269-5]
- 26. Enantioselective Construction of Fluorinated Tertiary ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12584140/]
- 27. Enantioselective Synthesis of Fluorinated Indolizidinone ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10186376/]
- 28. Data for Photoenzymatic Asymmetric Hydroamination for ... [https://experts.illinois.edu/en/datasets/data-for-photoenzymatic-asymmetric-hydroamination-for-chiral-alky/]
- 29. Photocatalytic Anti-Markovnikov Hydroamination of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10589911/]
- 30. Intermolecular Anti-Markovnikov Hydroamination of Alkenes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11804897/]
- 31. Genetically encoded artificial photoenzymes for ... [https://pubmed.ncbi.nlm.nih.gov/41167845/]
- 32. Stereoselective Photoenzymatic Hydroarylation for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12616453/]
- 33. Exploring the Increased Activity of the Blue Light ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12012757/]
- 34. The Impact of Low-Level Laser Irradiation on the Activity ... [https://www.mdpi.com/2304-6732/12/8/774]
- 35. Photoenzymatic enantioselective synthesis of fluorinated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12214493/]
- 36. Cofactor-independent photo-enzymatic reductions with ... [https://www.nature.com/articles/s41467-025-61908-6]
- 37. Continuous-flow chemoenzymatic enantioselective ... [https://www.sciencedirect.com/science/article/abs/pii/S2468823125002391]
- 38. Precise redesign for improving enzyme robustness based on ... [https://pubs.rsc.org/en/content/articlehtml/2024/sc/d4sc02058h]
- 39. Synthetic Biology, Directed Evolution, and the Rational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10401280/]
- 40. Directed Evolution Technology for Engineering Enzymes in ... [https://sequencing.roche.com/us/en/article-listing/directed-evolution.html]
- 41. Machine learning for enzyme engineering, selection and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8299298/]
- 42. Directed Evolution of High-Affinity Antibody Mimics Using ... [https://www.sciencedirect.com/science/article/pii/S1074552102001874]
- 43. Energy transfer-enabled enantioselective photocyclization ... [https://www.nature.com/articles/s41557-025-01857-1]
- 44. Stereoselective amino acid synthesis by synergistic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10444520/]
- 45. Transitioning enzyme catalysis towards photocatalysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12059584/]
- 46. Basics of Catalysts [https://chem.libretexts.org/Bookshelves/Inorganic_Chemistry/Supplemental_Modules_and_Websites_(Inorganic_Chemistry)/Catalysis/Basics_of_Catalysts]
- 47. 4.6 Catalysis – Inorganic Chemistry for Chemical Engineers [https://pressbooks.bccampus.ca/inorganicchemistrychem250/chapter/catalysis/]
- 48. One-pot chemo- and photo-enzymatic linear cascade processes [https://pubs.rsc.org/en/content/articlehtml/2024/cs/d3cs00595j]
- 49. How To Calculate Enantiomeric Excess: Learn Quickly [https://pharmaguru.co/enantiomeric-excess/]
- 50. Principles of green chemistry: building a sustainable future [https://link.springer.com/article/10.1007/s44371-025-00152-9]
- 51. a systematic review of early phase sustainability assessments ... [https://pubs.rsc.org/en/content/articlehtml/2025/gc/d5gc02565f]
- 52. 2025 Green Chemistry Challenge Award Winners [https://www.acs.org/green-chemistry-sustainability/funding-and-recognition/gcca/2025-awards.html]
- 53. Engineering a photoenzyme to use red light [https://www.sciencedirect.com/science/article/abs/pii/S2451929424004868]
- 54. recent advances in chemical synthesis using enzymes [https://www.sciencedirect.com/science/article/abs/pii/S1367593124001121]
- 55. Photoenzymatic Enantioselective Synthesis of Oxygen- ... [https://ui.adsabs.harvard.edu/abs/2023AngCh.135E1762Z/abstract]
- 56. Volume 8 Issue 11, November 2025 [https://www.nature.com/natcatal/volumes/8/issues/11]